Datta Sutapa, Mukhopadhyay Subhasis
Department of Biophysics, Molecular Biology and Bioinformatics and Distributed Information Centre for Bioinformatics, University of Calcutta, West Bengal, India.
Int J Nanomedicine. 2014 May 10;9:2225-39. doi: 10.2147/IJN.S57526. eCollection 2014.
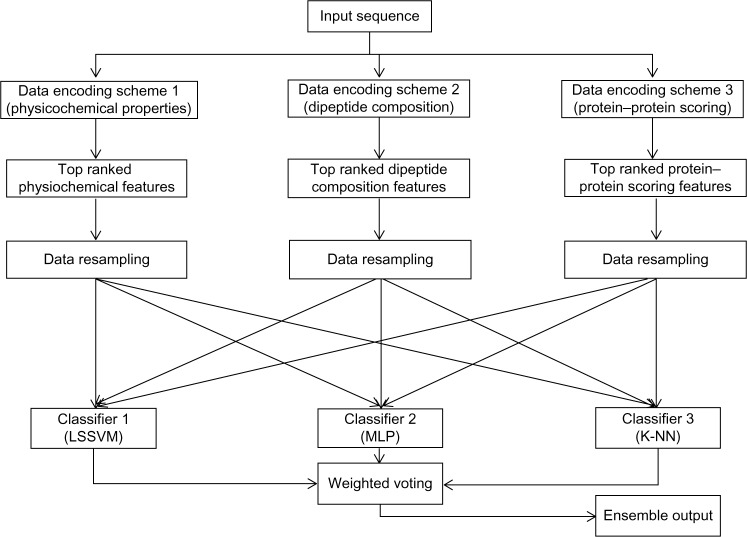
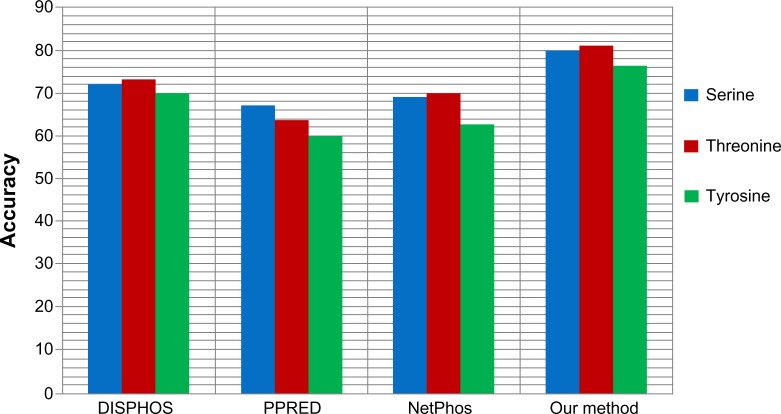
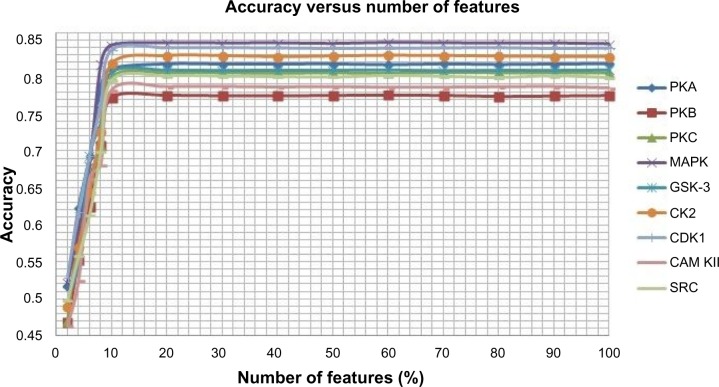
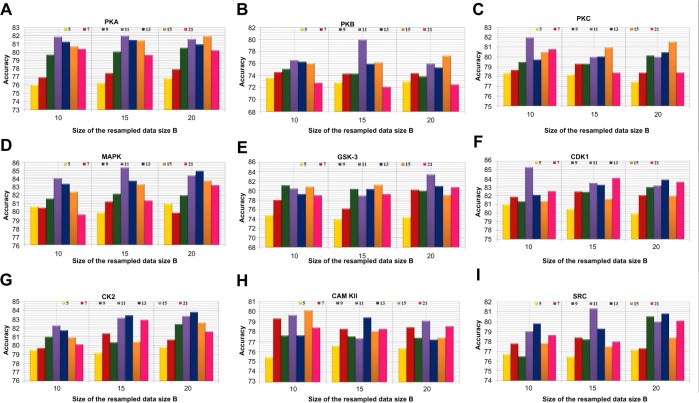
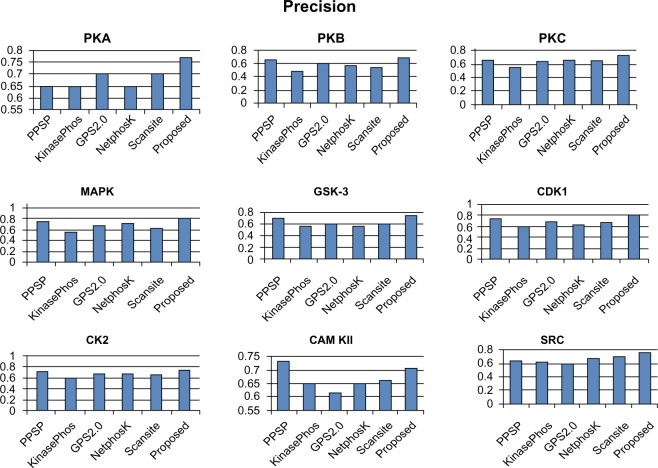
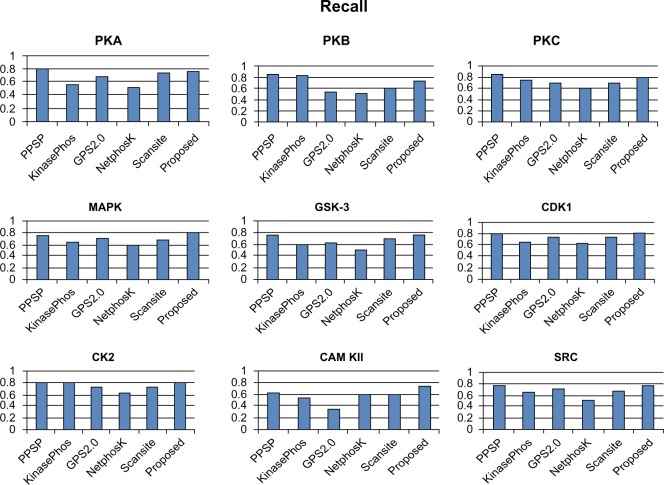
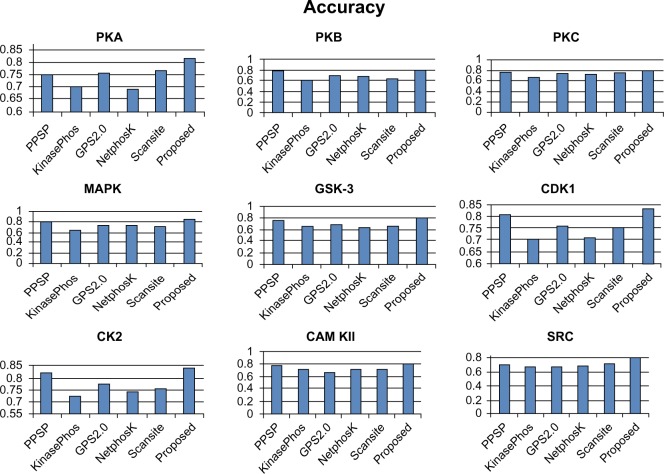
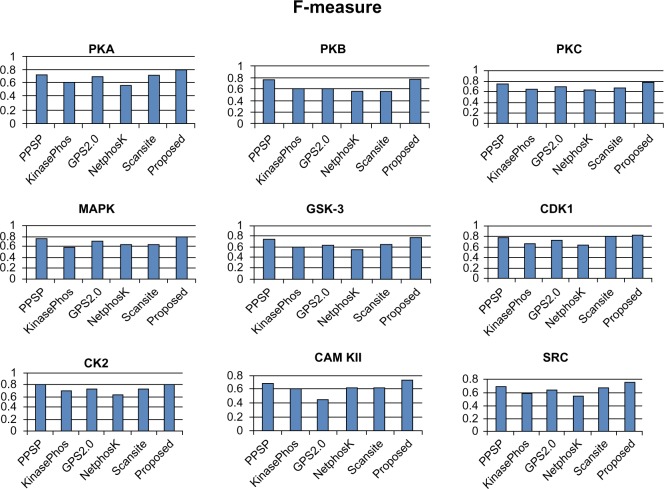
Protein phosphorylation is one of the most significant and well-studied post-translational modifications, and it plays an important role in various cellular processes. It has made a considerable impact in understanding the protein functions which are involved in revealing signal transductions and various diseases. The identification of kinase-specific phosphorylation sites has an important role in elucidating the mechanism of phosphorylation; however, experimental techniques for identifying phosphorylation sites are labor intensive and expensive. An exponentially increasing number of protein sequences generated by various laboratories across the globe require computer-aided procedures for reliably and quickly identifying the phosphorylation sites, opening a new horizon for in silico analysis. In this regard, we have introduced a novel ensemble method where we have selected three classifiers (least square support vector machine, multilayer perceptron, and k-Nearest Neighbor) and three different feature encoding parameters (dipeptide composition, physicochemical properties of amino acids, and protein-protein similarity score). Each of these classifiers is trained on each of the three different parameter systems. The final results of the ensemble method are obtained by fusing the results of all the classifiers by a weighted voting algorithm. Extensive experiments reveal that our proposed method can successfully predict phosphorylation sites in a kinase-specific manner and performs significantly better when compared with other existing phosphorylation site prediction methods.
蛋白质磷酸化是最重要且研究最深入的翻译后修饰之一,它在各种细胞过程中发挥着重要作用。在理解涉及揭示信号转导和各种疾病的蛋白质功能方面,它产生了相当大的影响。激酶特异性磷酸化位点的识别在阐明磷酸化机制中具有重要作用;然而,用于识别磷酸化位点的实验技术既费力又昂贵。全球各个实验室产生的蛋白质序列数量呈指数级增长,这就需要计算机辅助程序来可靠且快速地识别磷酸化位点,从而为计算机分析开辟了新的视野。在这方面,我们引入了一种新颖的集成方法,我们选择了三个分类器(最小二乘支持向量机、多层感知器和k近邻)以及三种不同的特征编码参数(二肽组成、氨基酸的物理化学性质和蛋白质 - 蛋白质相似性得分)。每个分类器在这三种不同的参数系统上进行训练。集成方法的最终结果是通过加权投票算法融合所有分类器的结果而获得的。大量实验表明,我们提出的方法能够以激酶特异性的方式成功预测磷酸化位点,并且与其他现有的磷酸化位点预测方法相比,性能有显著提升。