Authors' Affiliations: Cancer Vaccine Center; Department of Medical Oncology, Dana-Farber Cancer Institute; The Division of Allergy, Immunology, and Rheumatology, Department of Medicine, Massachusetts General Hospital; Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston; and Broad Institute of MIT and Harvard, Cambridge, MassachusettsAuthors' Affiliations: Cancer Vaccine Center; Department of Medical Oncology, Dana-Farber Cancer Institute; The Division of Allergy, Immunology, and Rheumatology, Department of Medicine, Massachusetts General Hospital; Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston; and Broad Institute of MIT and Harvard, Cambridge, MassachusettsAuthors' Affiliations: Cancer Vaccine Center; Department of Medical Oncology, Dana-Farber Cancer Institute; The Division of Allergy, Immunology, and Rheumatology, Department of Medicine, Massachusetts General Hospital; Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston; and Broad Institute of MIT and Harvard, Cambridge, Massachusetts.
Authors' Affiliations: Cancer Vaccine Center; Department of Medical Oncology, Dana-Farber Cancer Institute; The Division of Allergy, Immunology, and Rheumatology, Department of Medicine, Massachusetts General Hospital; Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston; and Broad Institute of MIT and Harvard, Cambridge, MassachusettsAuthors' Affiliations: Cancer Vaccine Center; Department of Medical Oncology, Dana-Farber Cancer Institute; The Division of Allergy, Immunology, and Rheumatology, Department of Medicine, Massachusetts General Hospital; Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston; and Broad Institute of MIT and Harvard, Cambridge, Massachusetts.
Cancer Immunol Res. 2014 Jun;2(6):522-9. doi: 10.1158/2326-6066.CIR-13-0227. Epub 2014 Mar 3.
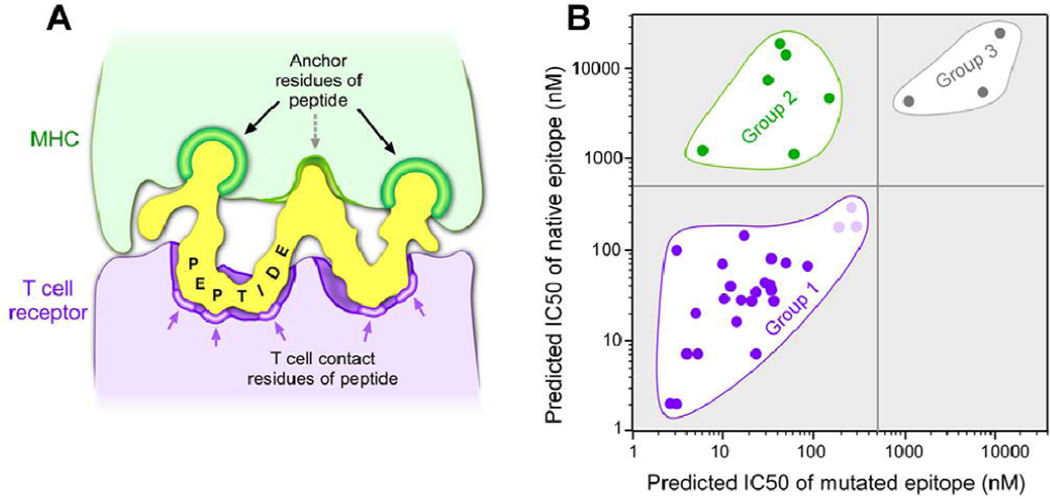
Cancer genome sequencing has enabled the rapid identification of the complete repertoire of coding sequence mutations within a patient's tumor and facilitated their use as personalized immunogens. Although a variety of techniques are available to assist in the selection of mutation-defined epitopes to be included within the tumor vaccine, the ability of the peptide to bind to patient MHC is a key gateway to peptide presentation. With advances in the accuracy of predictive algorithms for MHC class I binding, choosing epitopes on the basis of predicted affinity provides a rapid and unbiased approach to epitope prioritization. We show herein the retrospective application of a prediction algorithm to a large set of bona fide T cell-defined mutated human tumor antigens that induced immune responses, most of which were associated with tumor regression or long-term disease stability. The results support the application of this approach for epitope selection and reveal informative features of these naturally occurring epitopes to aid in epitope prioritization for use in tumor vaccines.
癌症基因组测序使我们能够快速识别患者肿瘤中编码序列突变的完整组合,并将其用作个性化免疫原。虽然有多种技术可用于辅助选择突变定义的表位,以包含在肿瘤疫苗中,但肽与患者 MHC 的结合能力是肽呈递的关键途径。随着 MHC 类 I 结合预测算法准确性的提高,基于预测亲和力选择表位为表位优先级排序提供了一种快速且无偏倚的方法。我们在此回顾性地将一种预测算法应用于一大组真正的 T 细胞定义的突变人类肿瘤抗原,这些抗原诱导了免疫反应,其中大多数与肿瘤消退或长期疾病稳定相关。这些结果支持了该方法在表位选择中的应用,并揭示了这些天然存在的表位的信息特征,以帮助对肿瘤疫苗中使用的表位进行优先级排序。