Giallonardo Francesca Di, Töpfer Armin, Rey Melanie, Prabhakaran Sandhya, Duport Yannick, Leemann Christine, Schmutz Stefan, Campbell Nottania K, Joos Beda, Lecca Maria Rita, Patrignani Andrea, Däumer Martin, Beisel Christian, Rusert Peter, Trkola Alexandra, Günthard Huldrych F, Roth Volker, Beerenwinkel Niko, Metzner Karin J
Division of Infectious Diseases and Hospital Epidemiology, University Hospital Zurich, University of Zurich, 8091 Zurich, Switzerland Life Science Zurich Graduate School, University of Zurich, 8057 Zurich, Switzerland.
Department of Biosystems Science and Engineering, ETH Zurich, 4058 Basel, Switzerland SIB Swiss Institute of Bioinformatics, 4058 Basel, Switzerland.
Nucleic Acids Res. 2014 Aug;42(14):e115. doi: 10.1093/nar/gku537. Epub 2014 Jun 27.
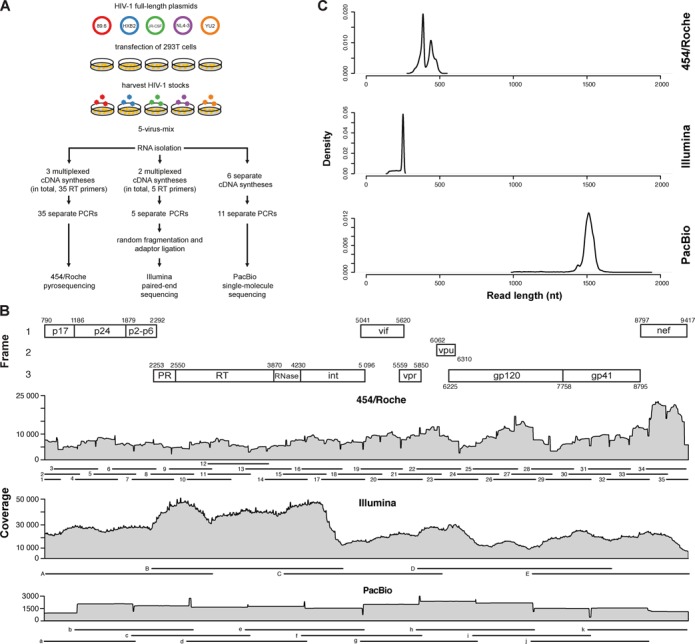
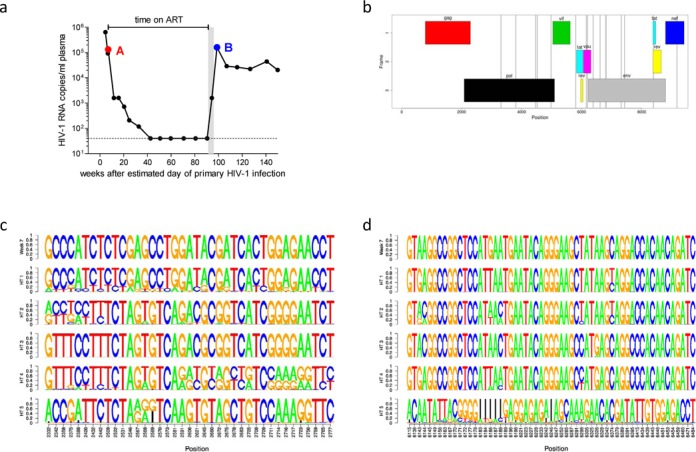
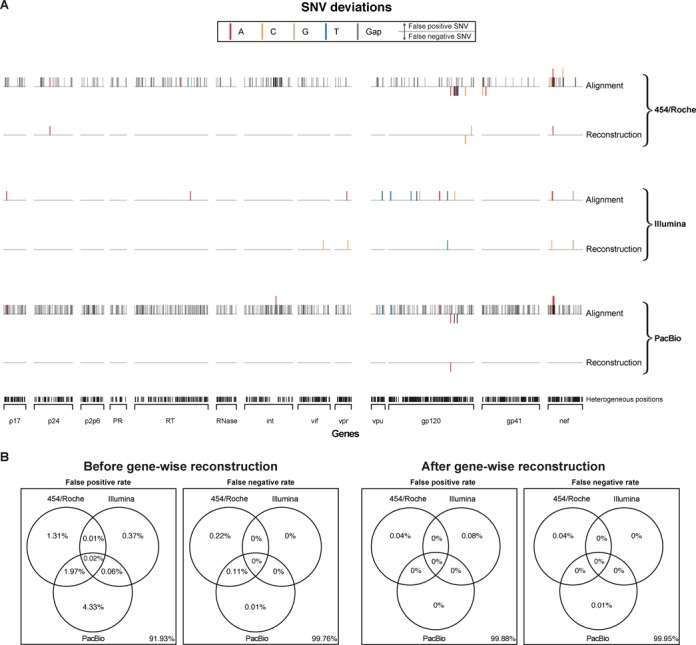
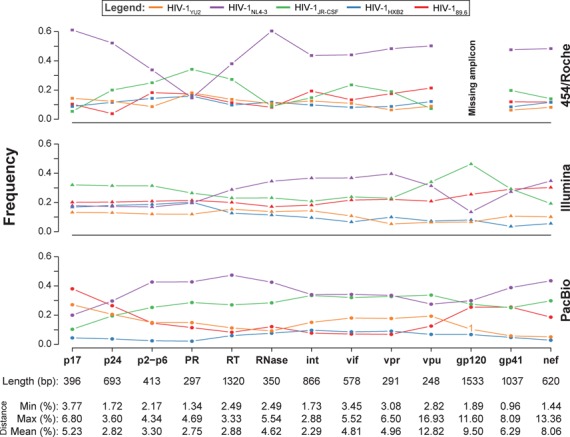
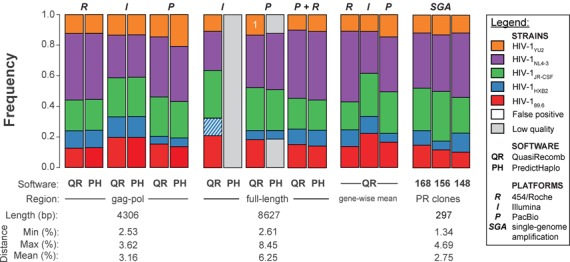
Next-generation sequencing (NGS) technologies enable new insights into the diversity of virus populations within their hosts. Diversity estimation is currently restricted to single-nucleotide variants or to local fragments of no more than a few hundred nucleotides defined by the length of sequence reads. To study complex heterogeneous virus populations comprehensively, novel methods are required that allow for complete reconstruction of the individual viral haplotypes. Here, we show that assembly of whole viral genomes of ∼8600 nucleotides length is feasible from mixtures of heterogeneous HIV-1 strains derived from defined combinations of cloned virus strains and from clinical samples of an HIV-1 superinfected individual. Haplotype reconstruction was achieved using optimized experimental protocols and computational methods for amplification, sequencing and assembly. We comparatively assessed the performance of the three NGS platforms 454 Life Sciences/Roche, Illumina and Pacific Biosciences for this task. Our results prove and delineate the feasibility of NGS-based full-length viral haplotype reconstruction and provide new tools for studying evolution and pathogenesis of viruses.
新一代测序(NGS)技术使人们能够对宿主体内病毒群体的多样性有新的认识。目前,多样性估计仅限于单核苷酸变异或由测序读长定义的不超过几百个核苷酸的局部片段。为了全面研究复杂的异质病毒群体,需要新的方法来实现对单个病毒单倍型的完整重建。在这里,我们表明,从来自克隆病毒株的特定组合以及一名HIV-1双重感染个体的临床样本中获得的异质HIV-1毒株混合物中,组装长度约为8600个核苷酸的完整病毒基因组是可行的。通过优化的实验方案以及用于扩增、测序和组装的计算方法实现了单倍型重建。我们比较评估了454生命科学公司/罗氏公司、Illumina和太平洋生物科学公司这三个NGS平台在此任务中的性能。我们的结果证明并描述了基于NGS的全长病毒单倍型重建的可行性,并为研究病毒的进化和发病机制提供了新工具。