Rumaney Maryam Bibi, Ngo Bitoungui Valentina Josiane, Vorster Anna Alvera, Ramesar Raj, Kengne Andre Pascal, Ngogang Jeanne, Wonkam Ambroise
Division of Human Genetics, Institute of Infectious Disease and Molecular Medicine (IDM), Faculty of Health Sciences, University of Cape Town (UCT), Cape Town, Republic of South Africa.
Department of Microbiology, Parasitology and Haematology, Faculty of Medicine and Biomedical Sciences, University of Yaoundé 1, Yaoundé, Cameroon.
PLoS One. 2014 Jun 30;9(6):e100516. doi: 10.1371/journal.pone.0100516. eCollection 2014.
Co-inheritance of α-thalassemia was reported to be associated with a delayed age of disease onset among Cameroonian Sickle Cell Anemia (SCA) patients. The present study aimed to explore the correlation between α-thalassemia, hematological indices, and clinical events in these patients.
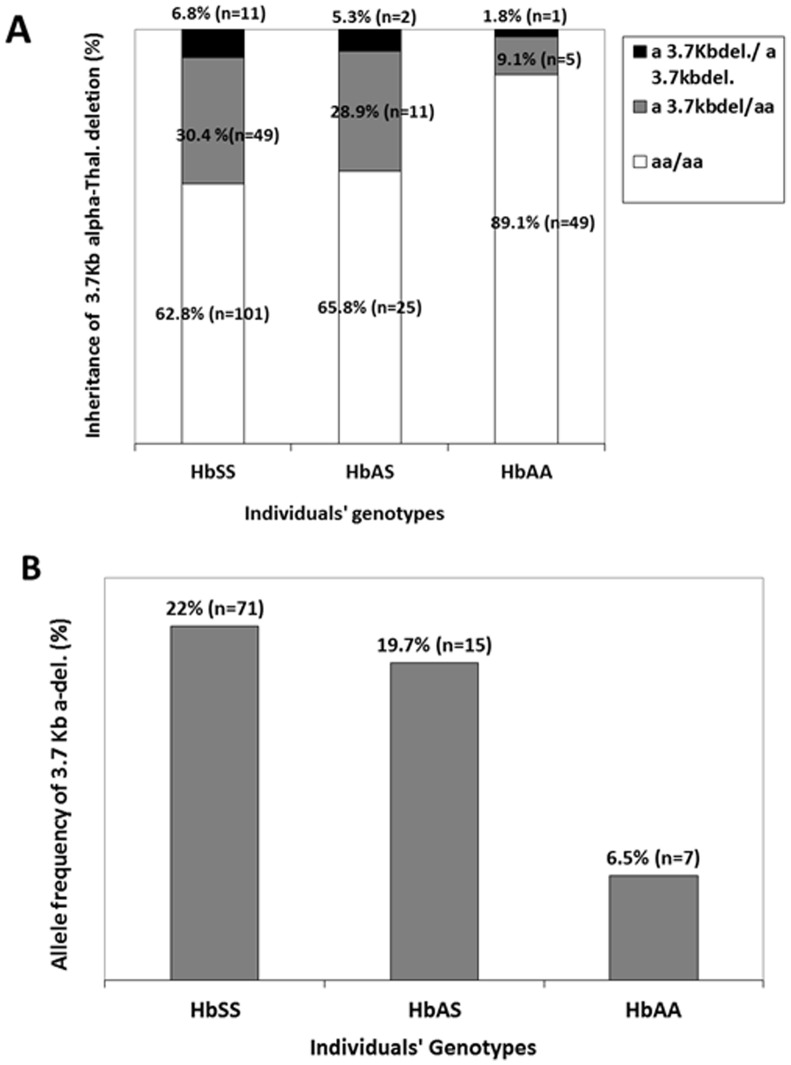
We studied 161 Cameroonian SCA patients and 103 controls (59.1% HbAA) with median ages of 17.5 and 23 years. RFLP-PCR was used to confirm SCA genotype and to describe haplotypes in the HBB-like genes cluster. Multiplex Gap-PCR was performed to investigate the 3.7 kb α-globin gene deletions. SNaPshot PCR, capillary electrophoresis and cycle sequencing were used for the genotyping of 10 SNPs in BCL11A, HMIP1/2, OR51B5/6 and HBG loci, known to influence HbF levels. Generalised linear regression models adjusted for age, sex and SNPs genotypes was used to investigate effects of α-thalassemia on clinical and hematological indices. The median rate of vaso-occlusive painful crisis and hospitalisations was two and one per year, respectively. Stroke was reported in eight cases (7.4%). Benin haplotype was the most prevalent (66.3%; n = 208 chromosomes). Among patients, 37.3% (n = 60) had at least one 3.7 kb deletion, compared to 10.9% (n = 6) among HbAA controls (p<0.001). Among patients, the median RBC count increased with the number of 3.7 kb deletions [2.6, 3.0 and 3.4 million/dl, with no, one and two deletions (p = 0.01)]. The median MCV decreased with the number of 3.7 kb deletion [86, 80, and 68fl, with no, one and two deletions (p<0.0001)], as well as median WBC counts [13.2, 10.5 and 9.8×109/L (p<0.0001. The co-inheritance of α-thalassemia was associated with lower consultations rate (p = 0.038).
The co-inheritance of α-thalassemia and SCA is associated with improved hematological indices, and lower consultations rate in this group of patients. This could possibly improve their survival and explain the higher proportion of α-thalassemia among patients than controls.
据报道,在喀麦隆镰状细胞贫血(SCA)患者中,α地中海贫血的共同遗传与疾病发病年龄延迟有关。本研究旨在探讨这些患者中α地中海贫血、血液学指标和临床事件之间的相关性。
我们研究了161例喀麦隆SCA患者和103例对照(59.1%为HbAA),中位年龄分别为17.5岁和23岁。采用限制性片段长度多态性聚合酶链反应(RFLP-PCR)来确认SCA基因型,并描述HBB样基因簇中的单倍型。进行多重缺口聚合酶链反应(Multiplex Gap-PCR)以检测3.7 kbα珠蛋白基因缺失。使用单核苷酸多态性分型技术(SNaPshot PCR)、毛细管电泳和循环测序对BCL11A、HMIP1/2、OR51B5/6和HBG基因座中的10个单核苷酸多态性(SNP)进行基因分型,这些基因座已知会影响胎儿血红蛋白(HbF)水平。采用针对年龄、性别和SNP基因型进行调整的广义线性回归模型来研究α地中海贫血对临床和血液学指标的影响。血管闭塞性疼痛危机和住院的中位发生率分别为每年两次和一次。有8例(7.4%)报告发生中风。贝宁单倍型最为常见(66.3%;n = 208条染色体)。在患者中,37.3%(n = 60)至少有一个3.7 kb缺失,而在HbAA对照中这一比例为10.9%(n = 6)(p<0.001)。在患者中,红细胞计数中位数随3.7 kb缺失数量的增加而升高[无缺失、有一个缺失和有两个缺失时分别为260万/dl、300万/dl和340万/dl(p = 0.01)]。平均红细胞体积(MCV)中位数随3.7 kb缺失数量的增加而降低[无缺失、有一个缺失和有两个缺失时分别为86fl、80fl和68fl(p<0.0001)],白细胞计数中位数也降低[分别为13.2×10⁹/L、10.5×10⁹/L和9.8×10⁹/L(p<0.0001)]。α地中海贫血的共同遗传与较低的就诊率相关(p = 0.038)。
α地中海贫血与SCA的共同遗传与血液学指标改善及该组患者较低的就诊率相关。这可能会改善他们的生存状况,并解释患者中α地中海贫血比例高于对照的原因。