Section on Statistical Genetics, Department of Biostatistics, University of Alabama at Birmingham Birmingham, AL, USA.
Section on Statistical Genetics, Department of Biostatistics, University of Alabama at Birmingham Birmingham, AL, USA ; Queensland Brain Institute, The University of Queensland St. Lucia, QLD, Australia.
Front Genet. 2014 Aug 12;5:267. doi: 10.3389/fgene.2014.00267. eCollection 2014.
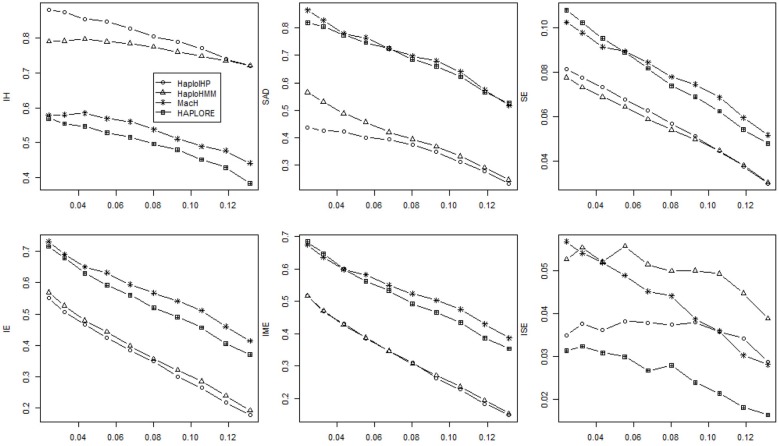
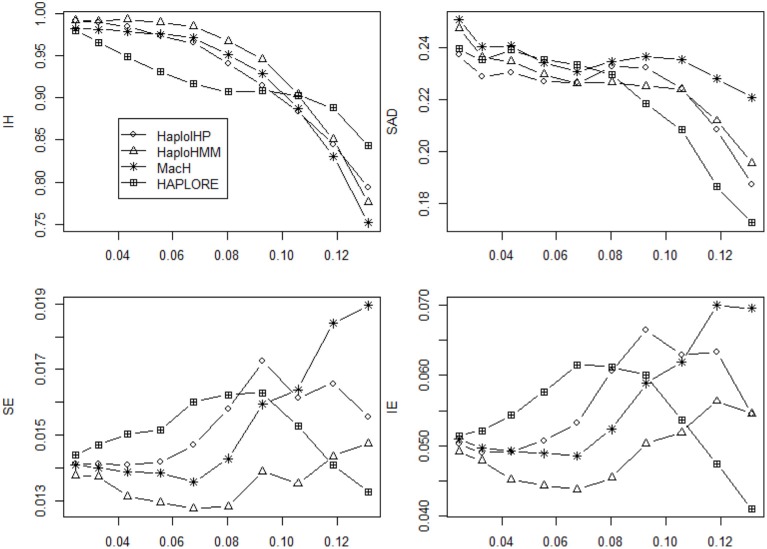
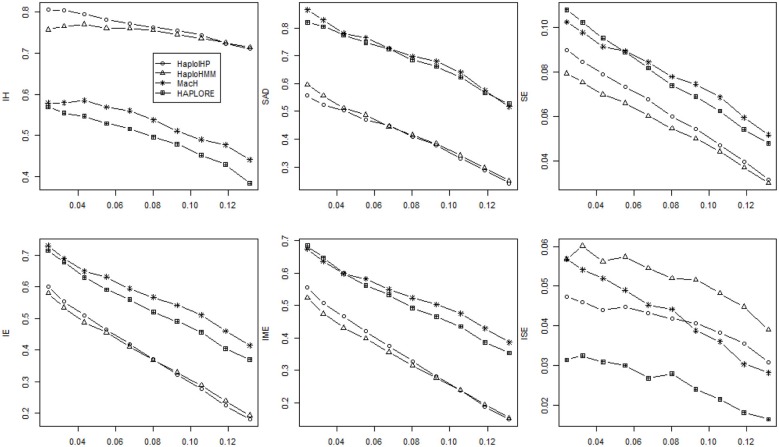
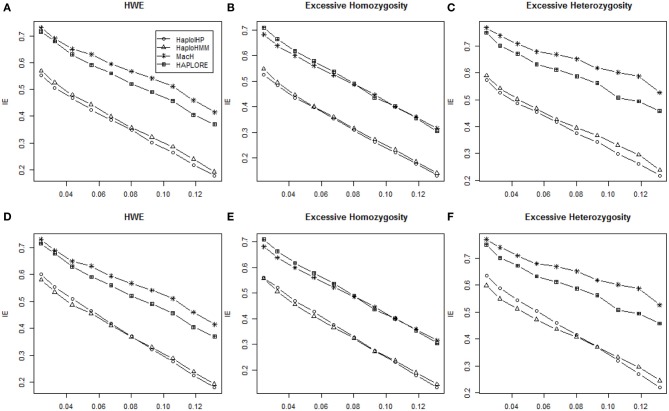
The majority of killer cell immunoglobin-like receptor (KIR) genes are detected as either present or absent using locus-specific genotyping technology. Ambiguity arises from the presence of a specific KIR gene since the exact copy number (one or two) of that gene is unknown. Therefore, haplotype inference for these genes is becoming more challenging due to such large portion of missing information. Meantime, many haplotypes and partial haplotype patterns have been previously identified due to tight linkage disequilibrium (LD) among these clustered genes thus can be incorporated to facilitate haplotype inference. In this paper, we developed a hidden Markov model (HMM) based method that can incorporate identified haplotypes or partial haplotype patterns for haplotype inference from present-absent data of clustered genes (e.g., KIR genes). We compared its performance with an expectation maximization (EM) based method previously developed in terms of haplotype assignments and haplotype frequency estimation through extensive simulations for KIR genes. The simulation results showed that the new HMM based method outperformed the previous method when some incorrect haplotypes were included as identified haplotypes and/or the standard deviation of haplotype frequencies were small. We also compared the performance of our method with two methods that do not use previously identified haplotypes and haplotype patterns, including an EM based method, HPALORE, and a HMM based method, MaCH. Our simulation results showed that the incorporation of identified haplotypes and partial haplotype patterns can improve accuracy for haplotype inference. The new software package HaploHMM is available and can be downloaded at http://www.soph.uab.edu/ssg/files/People/KZhang/HaploHMM/haplohmm-index.html.
大多数杀伤细胞免疫球蛋白样受体(KIR)基因使用基因座特异性基因分型技术检测为存在或不存在。由于该基因的确切拷贝数(一个或两个)未知,因此存在特定的 KIR 基因时会出现歧义。因此,由于这种大量缺失信息,这些基因的单体型推断变得更加具有挑战性。同时,由于这些聚集的基因之间存在紧密的连锁不平衡(LD),因此已经确定了许多单体型和部分单体型模式,从而可以将其纳入以促进单体型推断。在本文中,我们开发了一种基于隐马尔可夫模型(HMM)的方法,该方法可以从聚类基因(例如 KIR 基因)的存在-缺失数据中结合已识别的单体型或部分单体型模式进行单体型推断。我们通过对 KIR 基因进行广泛的模拟,比较了其与先前基于期望最大化(EM)的方法在单体型分配和单体型频率估计方面的性能。模拟结果表明,当将一些不正确的单体型作为已识别的单体型包含在内和/或单体型频率的标准偏差较小时,新的基于 HMM 的方法优于先前的方法。我们还比较了我们的方法与不使用先前确定的单体型和单体型模式的两种方法的性能,包括基于 EM 的方法 HPALORE 和基于 HMM 的方法 MaCH。我们的模拟结果表明,结合已识别的单体型和部分单体型模式可以提高单体型推断的准确性。新的软件包 HaploHMM 可在以下网址获得并下载:http://www.soph.uab.edu/ssg/files/People/KZhang/HaploHMM/haplohmm-index.html。