Bone Robert N, Gai Ying, Magrioti Victoria, Kokotou Maroula G, Ali Tomader, Lei Xiaoyong, Tse Hubert M, Kokotos George, Ramanadham Sasanka
Department of Pathology, University of Alabama at Birmingham, Birmingham, AL Comprehensive Diabetes Center, University of Alabama at Birmingham, Birmingham, AL.
Comprehensive Diabetes Center, University of Alabama at Birmingham, Birmingham, AL Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL.
Diabetes. 2015 Feb;64(2):541-54. doi: 10.2337/db14-0097. Epub 2014 Sep 11.
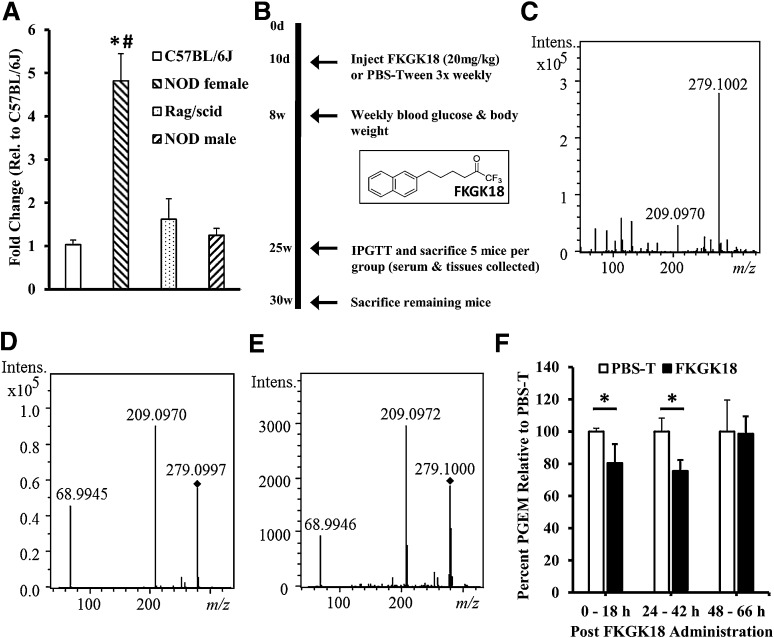
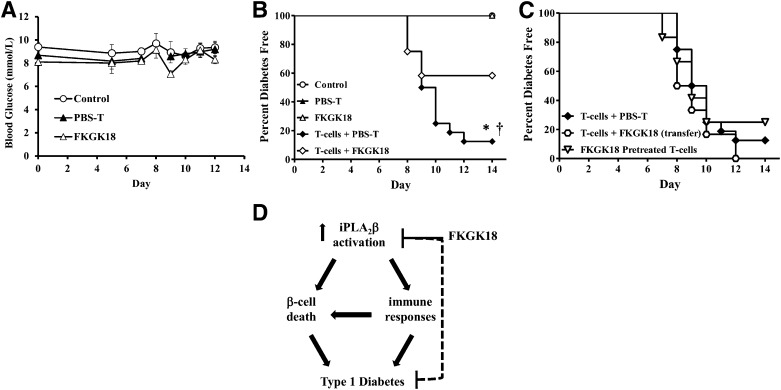
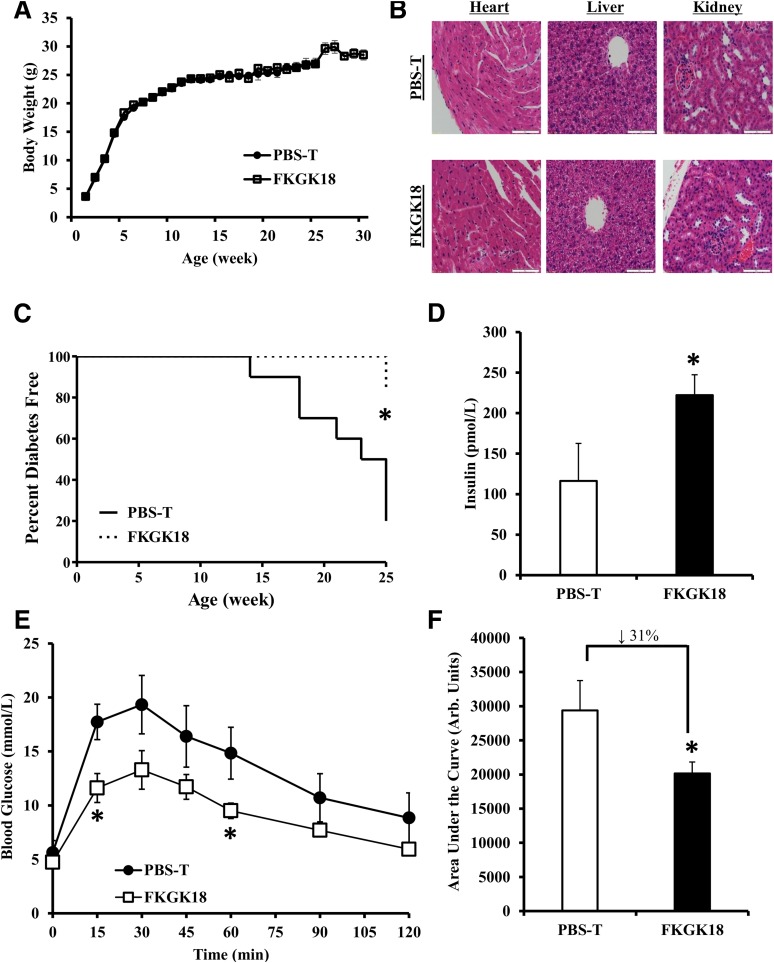
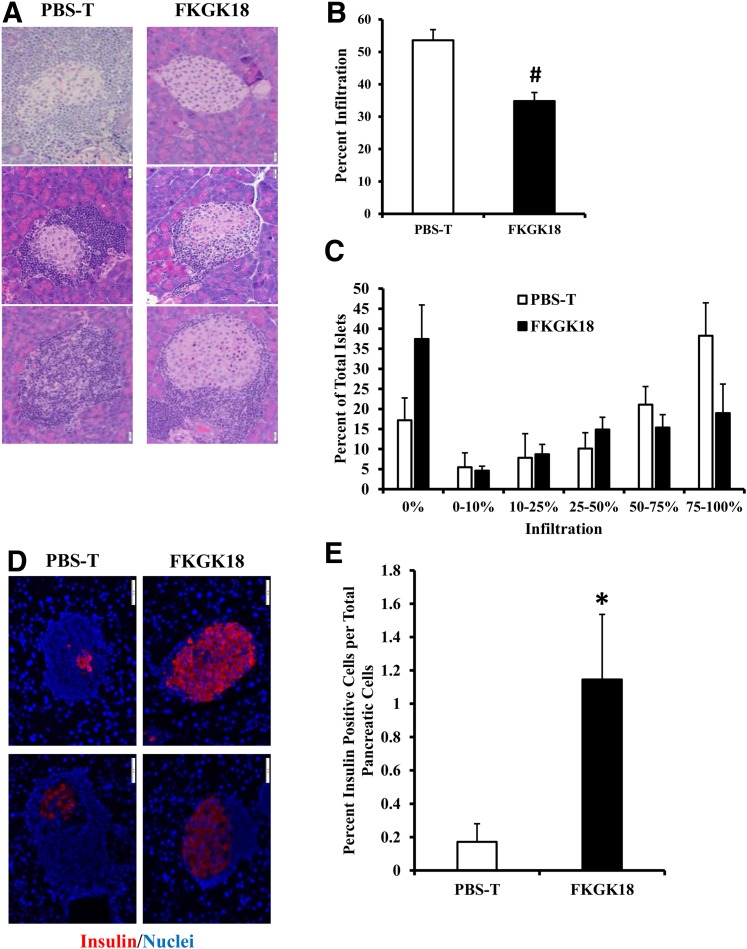
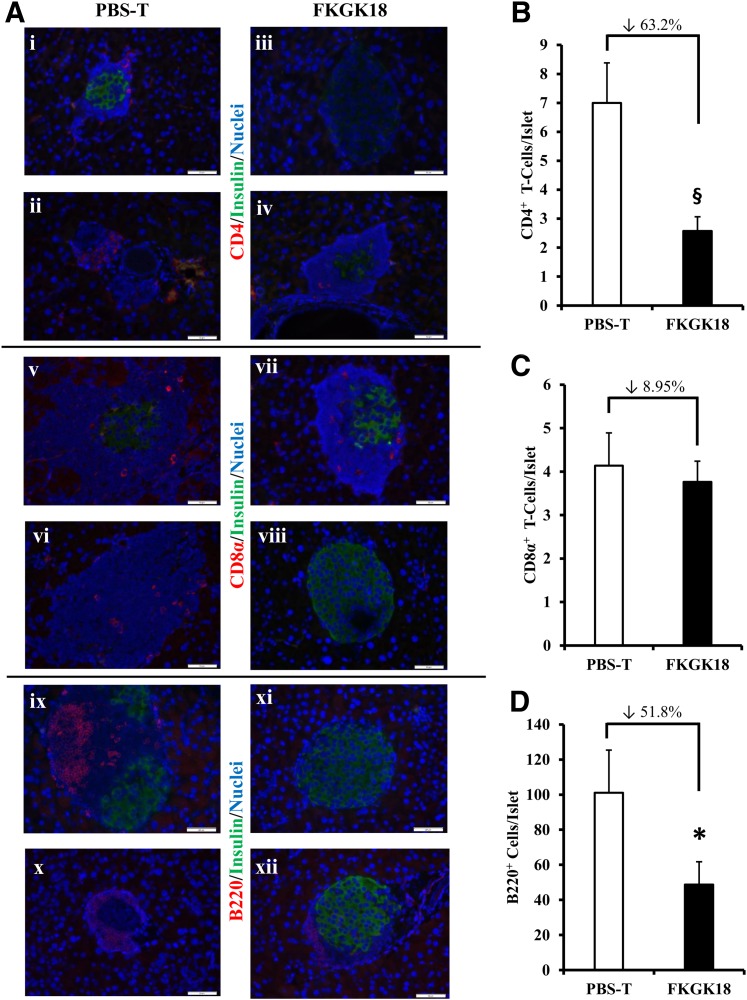
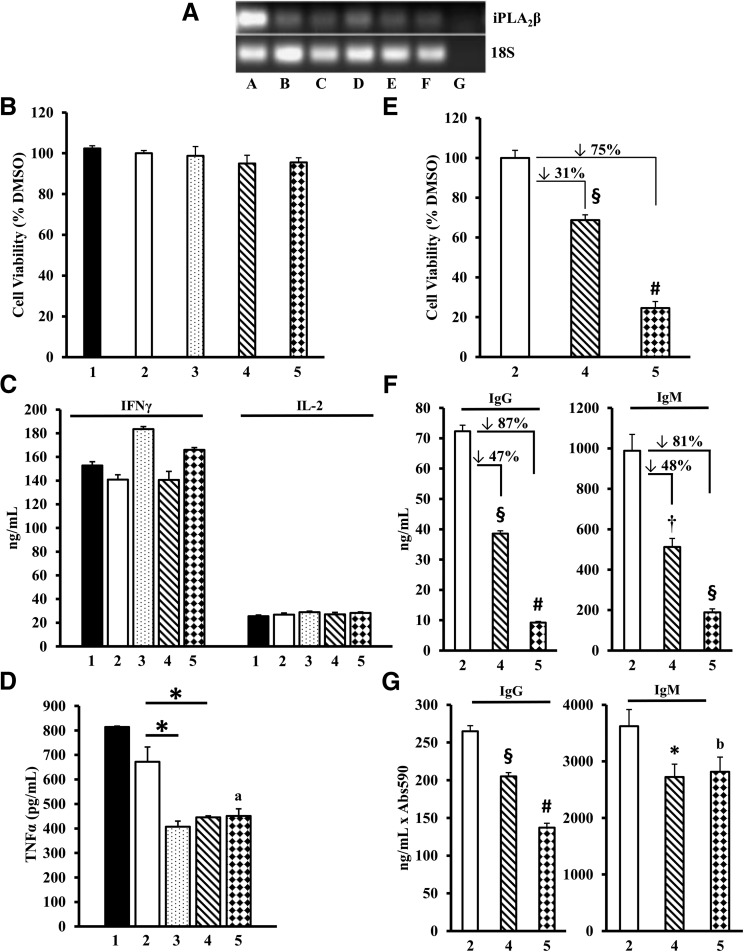
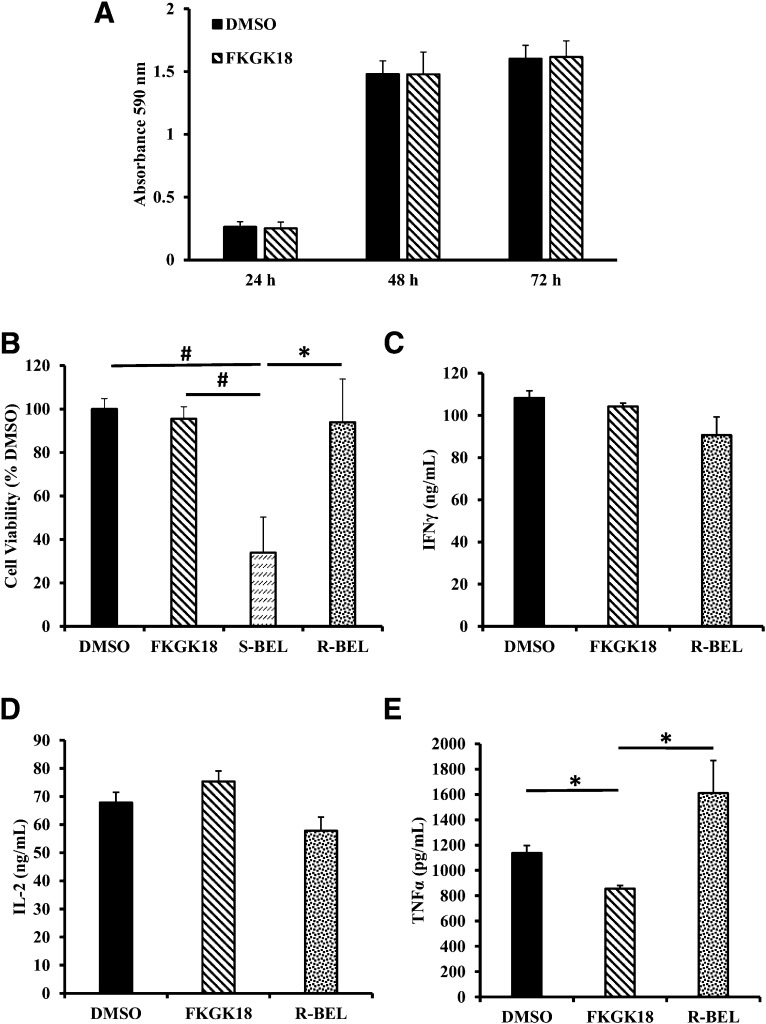
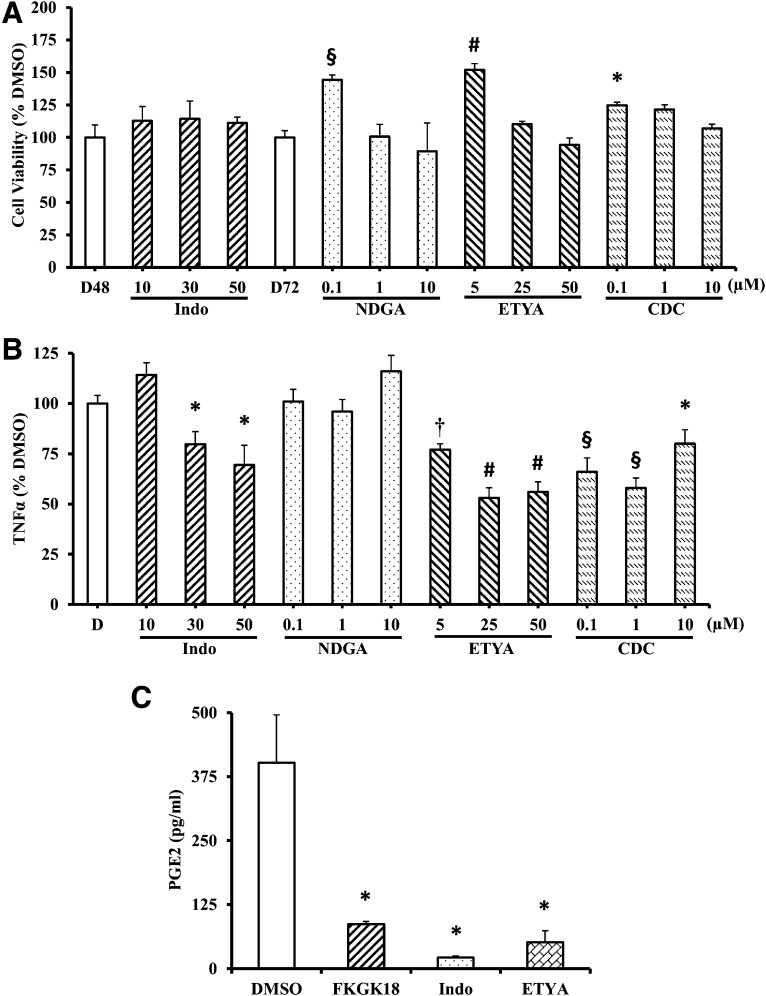
Autoimmune β-cell death leads to type 1 diabetes, and with findings that Ca(2+)-independent phospholipase A2β (iPLA2β) activation contributes to β-cell death, we assessed the effects of iPLA2β inhibition on diabetes development. Administration of FKGK18, a reversible iPLA2β inhibitor, to NOD female mice significantly reduced diabetes incidence in association with 1) reduced insulitis, reflected by reductions in CD4(+) T cells and B cells; 2) improved glucose homeostasis; 3) higher circulating insulin; and 4) β-cell preservation. Furthermore, FKGK18 inhibited production of tumor necrosis factor-α (TNF-α) from CD4(+) T cells and antibodies from B cells, suggesting modulation of immune cell responses by iPLA2β-derived products. Consistent with this, 1) adoptive transfer of diabetes by CD4(+) T cells to immunodeficient and diabetes-resistant NOD.scid mice was mitigated by FKGK18 pretreatment and 2) TNF-α production from CD4(+) T cells was reduced by inhibitors of cyclooxygenase and 12-lipoxygenase, which metabolize arachidonic acid to generate bioactive inflammatory eicosanoids. However, adoptive transfer of diabetes was not prevented when mice were administered FKGK18-pretreated T cells or when FKGK18 administration was initiated with T-cell transfer. The present observations suggest that iPLA2β-derived lipid signals modulate immune cell responses, raising the possibility that early inhibition of iPLA2β may be beneficial in ameliorating autoimmune destruction of β-cells and mitigating type 1 diabetes development.
自身免疫性β细胞死亡会导致1型糖尿病,鉴于有研究发现不依赖钙离子的磷脂酶A2β(iPLA2β)激活会导致β细胞死亡,我们评估了抑制iPLA2β对糖尿病发展的影响。给非肥胖糖尿病(NOD)雌性小鼠施用可逆性iPLA2β抑制剂FKGK18,可显著降低糖尿病发病率,这与以下几点相关:1)胰岛炎减轻,表现为CD4(+) T细胞和B细胞数量减少;2)葡萄糖稳态改善;3)循环胰岛素水平升高;4)β细胞得以保存。此外,FKGK18抑制了CD4(+) T细胞产生肿瘤坏死因子-α(TNF-α)以及B细胞产生抗体,这表明iPLA2β衍生产物可调节免疫细胞反应。与此相符的是,1)FKGK18预处理可减轻CD4(+) T细胞将糖尿病过继转移至免疫缺陷且抗糖尿病的NOD.scid小鼠的情况,2)环氧化酶和12-脂氧合酶抑制剂可减少CD4(+) T细胞产生TNF-α,这两种酶可将花生四烯酸代谢生成具有生物活性的炎性类二十烷酸。然而,当给小鼠施用经FKGK18预处理的T细胞时,或者在T细胞转移时开始施用FKGK18,糖尿病的过继转移并未得到预防。目前的观察结果表明,iPLA2β衍生的脂质信号可调节免疫细胞反应,这增加了早期抑制iPLA2β可能有利于改善β细胞的自身免疫性破坏并减轻1型糖尿病发展的可能性。