Fang Xuqian, Liu Xiangfan, Yao Ling, Chen Changqiang, Lin Jiafei, Ni Peihua, Zheng Xinmin, Fan Qishi
Department of Clinical Laboratory, Ruijin North Hospital, Ruijin Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, P. R. China.
Faculty of Medical Laboratory Science, Shanghai JiaoTong University School of Medicine, Shanghai, P. R. China.
PLoS One. 2014 Sep 16;9(9):e107134. doi: 10.1371/journal.pone.0107134. eCollection 2014.
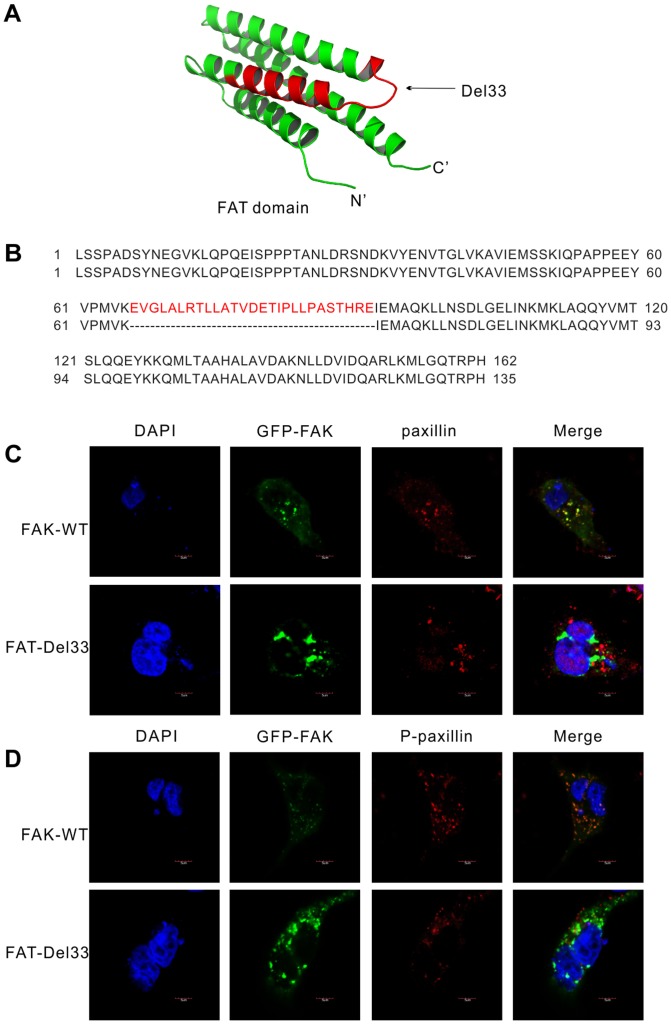
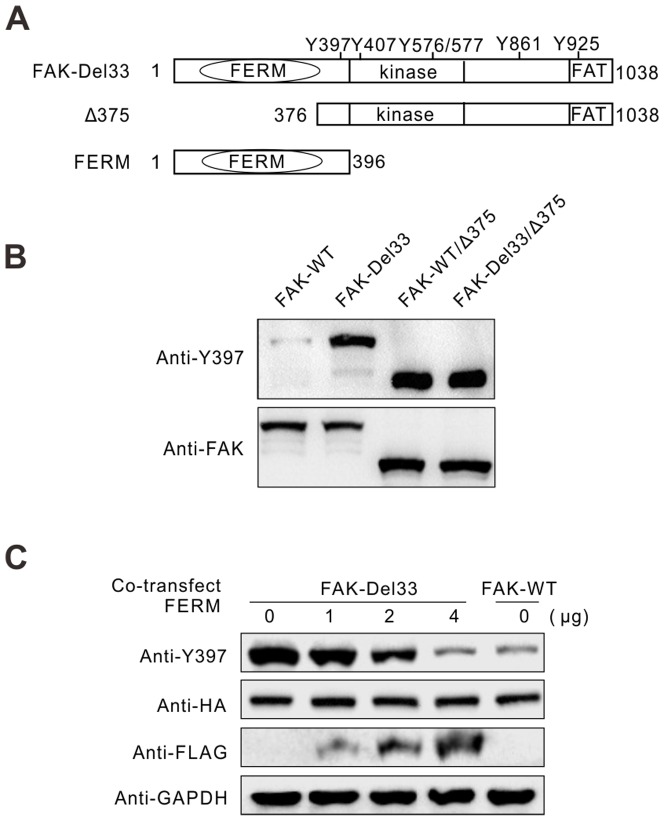
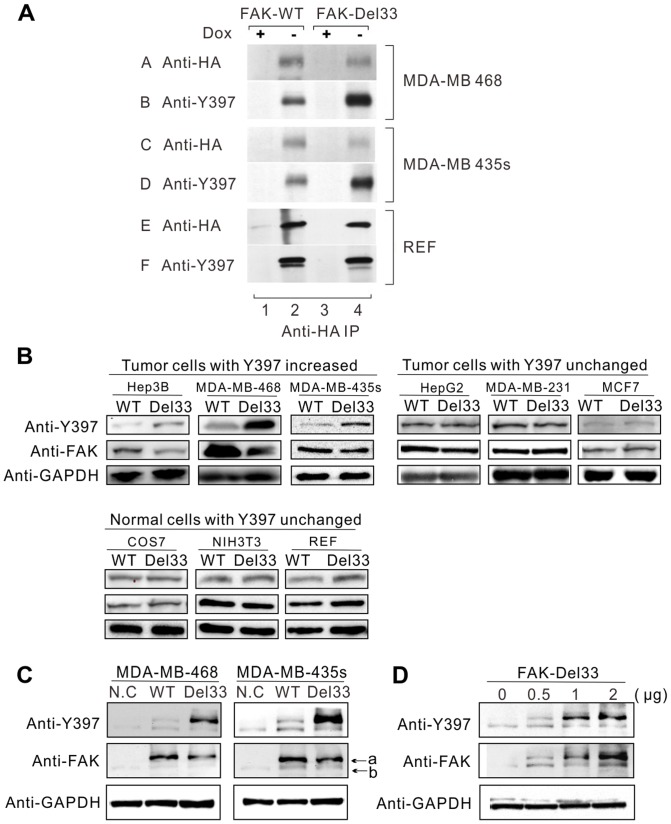
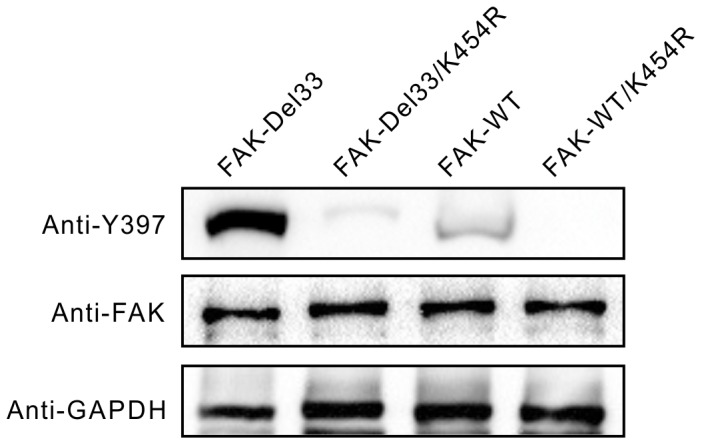
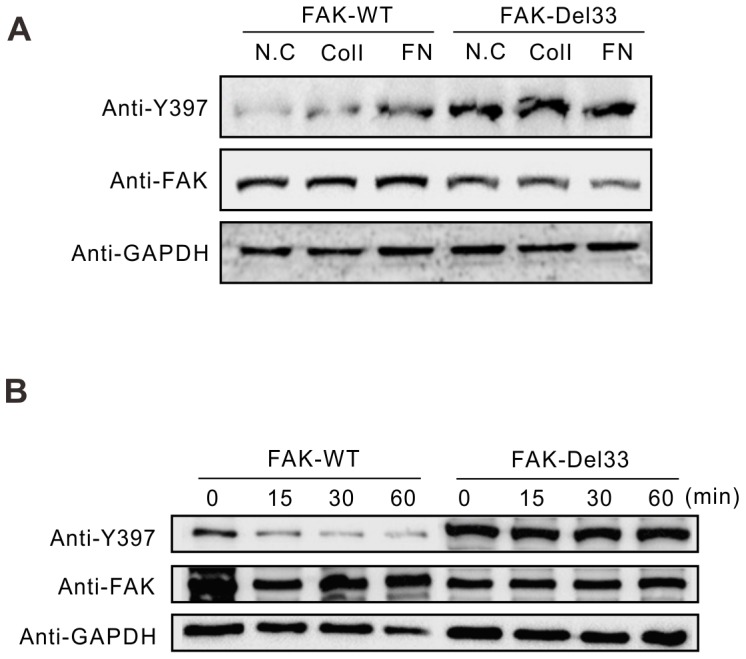
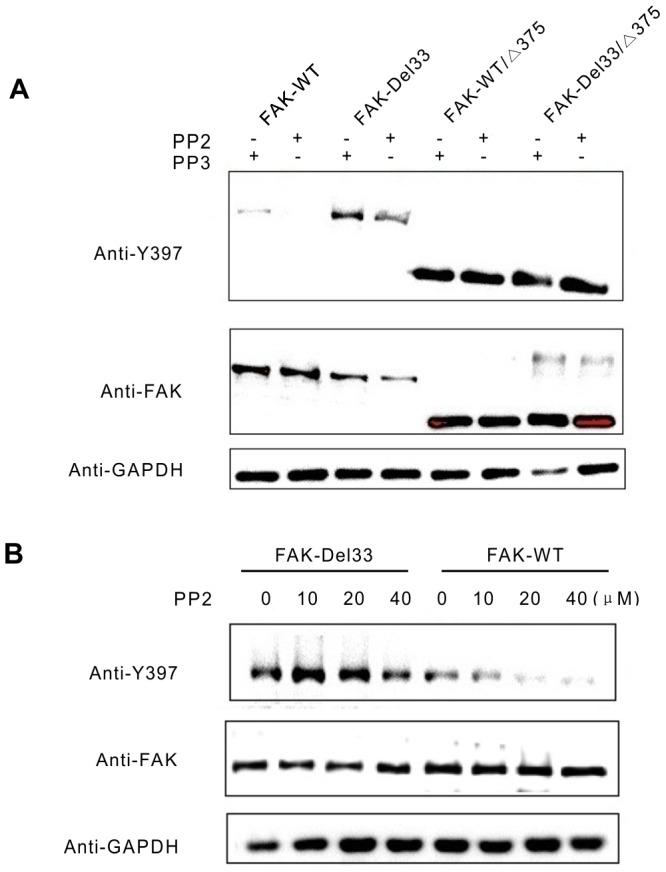
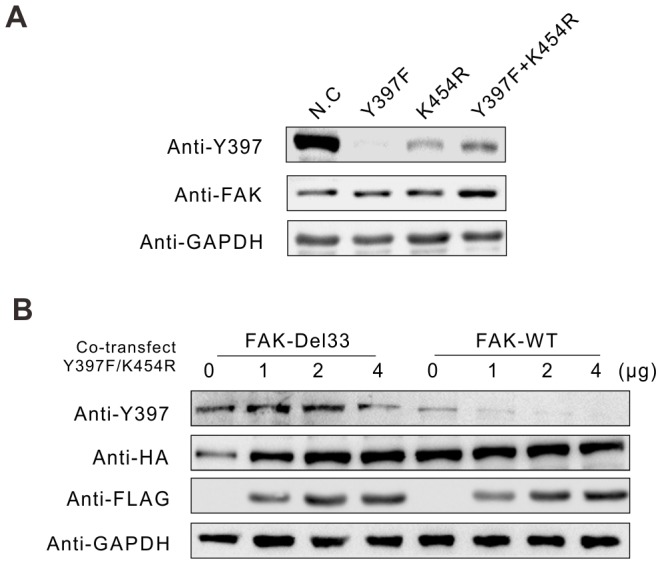
Mounting evidence suggests that the FAK N-terminal (FERM) domain controls FAK phosphorylation and function; however, little is known regarding the role of the C terminal (FAT) domain in FAK regulation. We identified a patient-derived FAK mutant, in which a 27-amino acid segment was deleted from the C-terminal FAT domain (named FAK-Del33). When FAK-Del33 was overexpressed in specific tumor cell lines, Y397 phosphorylation increased compared with that observed in cells expressing FAK-WT. Here, we attempt to unveil the mechanism of this increased phosphorylation. Using cell biology experiments, we show that FAK-Del33 is incapable of co-localizing with paxillin, and has constitutively high Y397 phosphorylation. With a kinase-dead mutation, it showed phosphorylation of FAK-Del33 has enhanced through auto-phosphorylation. It was also demonstrated that phosphorylation of FAK-Del33 is not Src dependent or enhanced intermolecular interactions, and that the hyperphosphorylation can be lowered using increasing amounts of transfected FERM domain. This result suggests that Del33 mutation disrupting of FAT's structural integrity and paxillin binding capacity leads to incapable of targeting Focal adhesions, but has gained the capacity for auto-phosphorylation in cis.
越来越多的证据表明,黏着斑激酶(FAK)的N端(FERM)结构域控制着FAK的磷酸化和功能;然而,关于C端(FAT)结构域在FAK调节中的作用却知之甚少。我们鉴定出一种源自患者的FAK突变体,其中C端FAT结构域缺失了一段27个氨基酸的片段(命名为FAK-Del33)。当FAK-Del33在特定肿瘤细胞系中过表达时,与表达野生型FAK(FAK-WT)的细胞相比,Y397位点的磷酸化增加。在此,我们试图揭示这种磷酸化增加的机制。通过细胞生物学实验,我们发现FAK-Del33无法与桩蛋白共定位,并且具有组成性的高Y397磷酸化。通过激酶失活突变,结果表明FAK-Del33的磷酸化通过自身磷酸化而增强。还证明了FAK-Del33的磷酸化不依赖于Src,也不是分子间相互作用增强导致的,并且使用增加量的转染FERM结构域可以降低过度磷酸化。这一结果表明,Del33突变破坏了FAT的结构完整性和桩蛋白结合能力,导致无法靶向粘着斑,但获得了顺式自身磷酸化的能力。