BMC Bioinformatics. 2014;15 Suppl 9(Suppl 9):S16. doi: 10.1186/1471-2105-15-S9-S16. Epub 2014 Sep 10.
It has recently become possible to rapidly and accurately detect epigenetic signatures in bacterial genomes using third generation sequencing data. Monitoring the speed at which a single polymerase inserts a base in the read strand enables one to infer whether a modification is present at that specific site on the template strand. These sites can be challenging to detect in the absence of high coverage and reliable reference genomes.
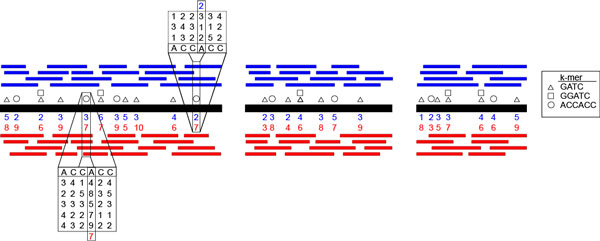
Here we provide a new method for detecting epigenetic motifs in bacteria on datasets with low-coverage, with incomplete references, and with mixed samples (i.e. metagenomic data). Our approach treats motif inference as a kmer comparison problem. First, genomes (or contigs) are deconstructed into kmers. Then, native genome-wide distributions of interpulse durations (IPDs) for kmers are compared with corresponding whole genome amplified (WGA, modification free) IPD distributions using log likelihood ratios. Finally, kmers are ranked and greedily selected by iteratively correcting for sequences within a particular kmer's neighborhood.
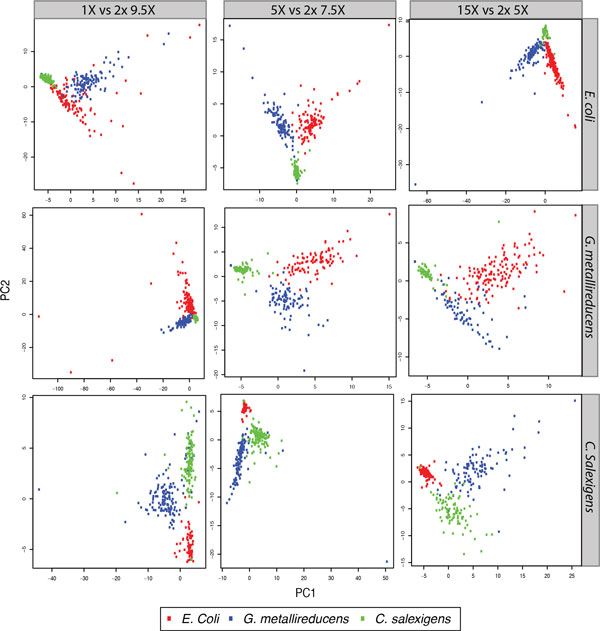
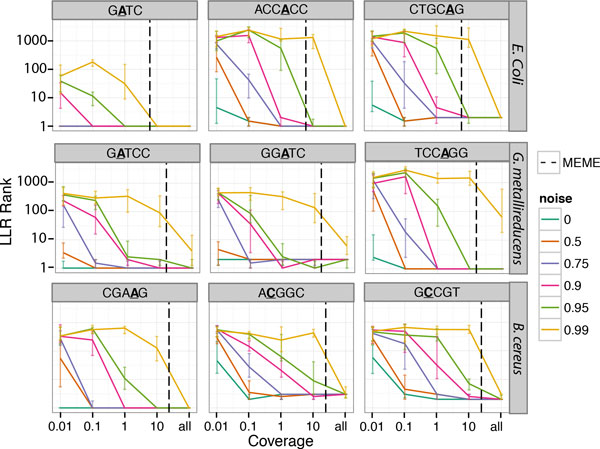
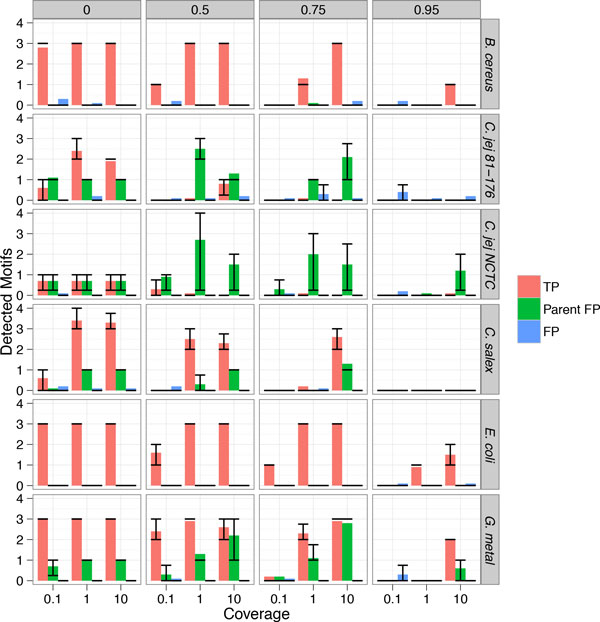
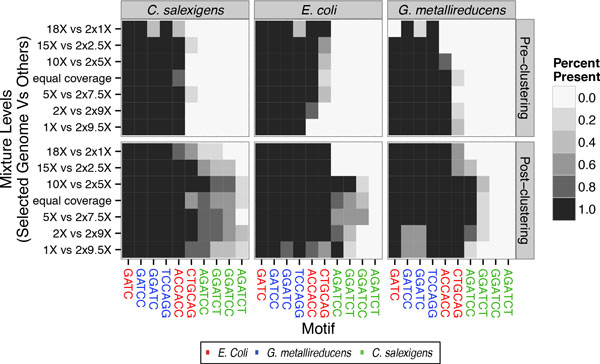
Our method can detect multiple types of modifications, even at very low-coverage and in the presence of mixed genomes. Additionally, we are able to predict modified motifs when genomes with "neighbor" modified motifs exist within the sample. Lastly, we show that these motifs can provide an alternative source of information by which to cluster metagenomics contigs and that iterative refinement on these clustered contigs can further improve both sensitivity and specificity of motif detection.
最近,使用第三代测序数据可以快速准确地检测细菌基因组中的表观遗传特征。监测单个聚合酶在读取链中插入碱基的速度,可以推断在模板链上的特定位置是否存在修饰。在没有高覆盖率和可靠参考基因组的情况下,这些位点很难检测到。
在这里,我们提供了一种新的方法,用于在低覆盖率、不完整的参考基因组和混合样本(即宏基因组数据)的数据集上检测细菌中的表观遗传基序。我们的方法将基序推断视为 kmer 比较问题。首先,将基因组(或 contigs)分解为 kmer。然后,使用对数似然比将 kmers 的内脉冲持续时间 (IPD) 的原始全基因组分布与相应的全基因组扩增 (WGA,无修饰) IPD 分布进行比较。最后,通过迭代校正特定 kmer 邻域内的序列对 kmers 进行排名和贪婪选择。
我们的方法可以检测多种类型的修饰,即使在覆盖率非常低和存在混合基因组的情况下也是如此。此外,我们能够在样本中存在具有“邻居”修饰基序的基因组时预测修饰基序。最后,我们表明,这些基序可以通过聚类宏基因组 contigs 提供替代信息源,并且对这些聚类 contigs 进行迭代细化可以进一步提高基序检测的灵敏度和特异性。