Numminen Elina, Chewapreecha Claire, Sirén Jukka, Turner Claudia, Turner Paul, Bentley Stephen D, Corander Jukka
Department of Mathematics and Statistics, University of Helsinki, PO Box 68, 00014 Helsinki, Finland
The Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton CB10 1SA, UK.
Proc Biol Sci. 2014 Nov 7;281(1794):20141324. doi: 10.1098/rspb.2014.1324.
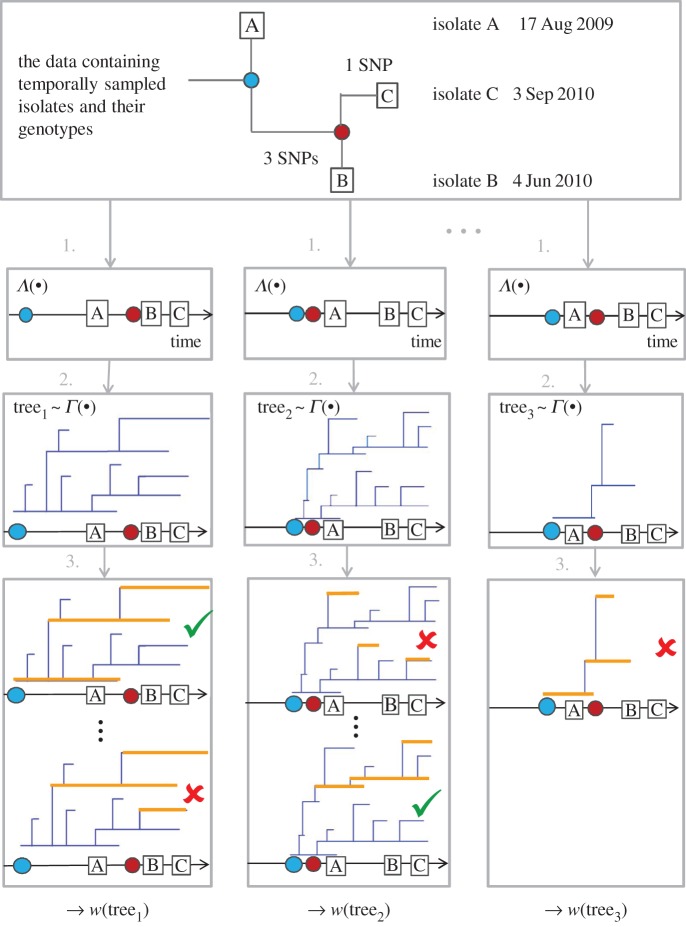
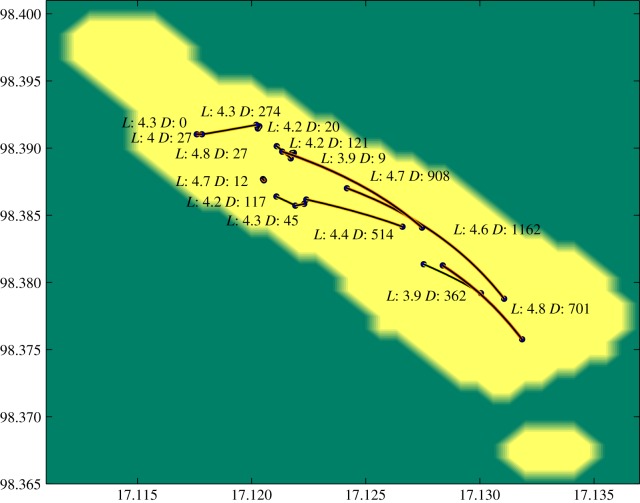
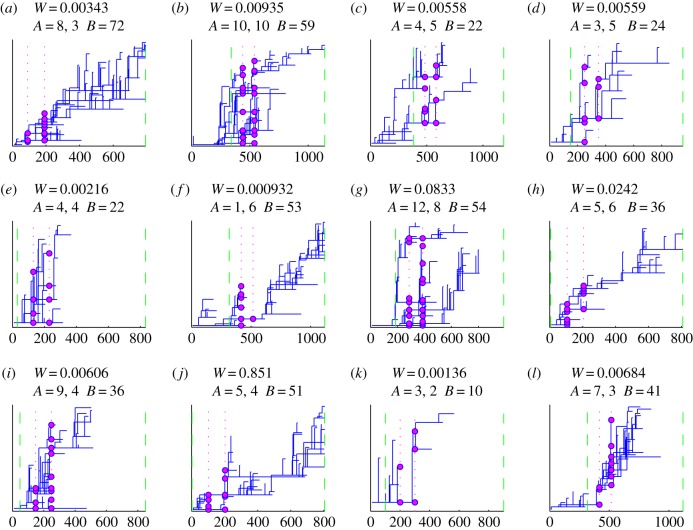
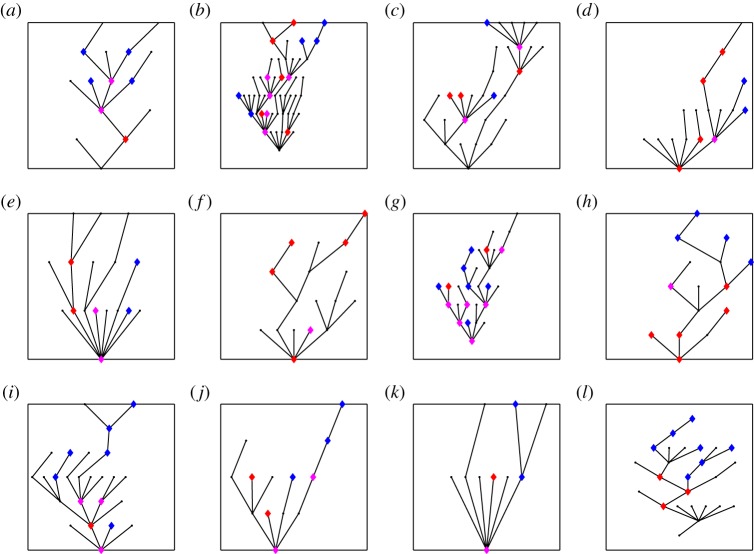
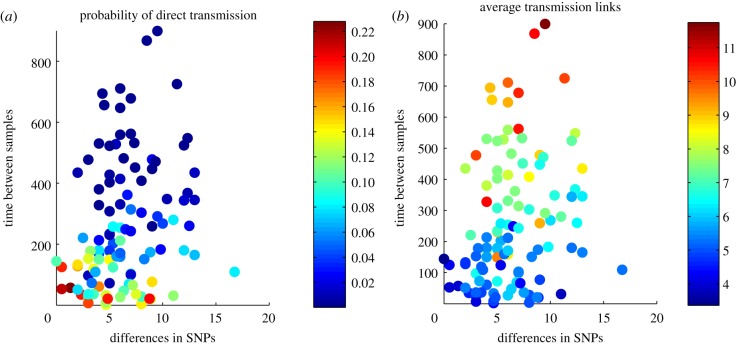
There has been growing interest in the statistics community to develop methods for inferring transmission pathways of infectious pathogens from molecular sequence data. For many datasets, the computational challenge lies in the huge dimension of the missing data. Here, we introduce an importance sampling scheme in which the transmission trees and phylogenies of pathogens are both sampled from reasonable importance distributions, alleviating the inference. Using this approach, arbitrary models of transmission could be considered, contrary to many earlier proposed methods. We illustrate the scheme by analysing transmissions of Streptococcus pneumoniae from household to household within a refugee camp, using data in which only a fraction of hosts is observed, but which is still rich enough to unravel the within-household transmission dynamics and pairs of households between whom transmission is plausible. We observe that while probability of direct transmission is low even for the most prominent cases of transmission, still those pairs of households are geographically much closer to each other than expected under random proximity.
统计学界对开发从分子序列数据推断传染病病原体传播途径的方法的兴趣与日俱增。对于许多数据集而言,计算挑战在于缺失数据的巨大维度。在此,我们引入一种重要性抽样方案,其中病原体的传播树和系统发育均从合理的重要性分布中进行抽样,从而减轻推断的难度。与许多早期提出的方法不同,使用这种方法可以考虑任意的传播模型。我们通过分析难民营内肺炎链球菌在家庭间的传播来说明该方案,所使用的数据中仅观察到一部分宿主,但这些数据仍足以揭示家庭内部的传播动态以及可能存在传播关系的家庭对。我们观察到,即使对于最显著的传播案例,直接传播的概率也很低,但这些有传播关系的家庭对在地理上彼此之间的距离仍比随机接近情况下预期的要近得多。