Wong C, Laddha S V, Tang L, Vosburgh E, Levine A J, Normant E, Sandy P, Harris C R, Chan C S, Xu E Y
Raymond and Beverly Sackler Foundation Laboratory, 195 Little Albany Street, New Brunswick, NJ 08901, USA.
1] Rutgers Cancer Institute of New Jersey, Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey, 195 Little Albany Street, New Brunswick, NJ 08901, USA [2] Department of Medicine, Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey, 195 Little Albany Street, New Brunswick, NJ 08901, USA [3] Center for Systems Biology, Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey, 195 Little Albany Street, New Brunswick, NJ 08901, USA.
Cell Death Dis. 2014 Oct 9;5(10):e1450. doi: 10.1038/cddis.2014.396.
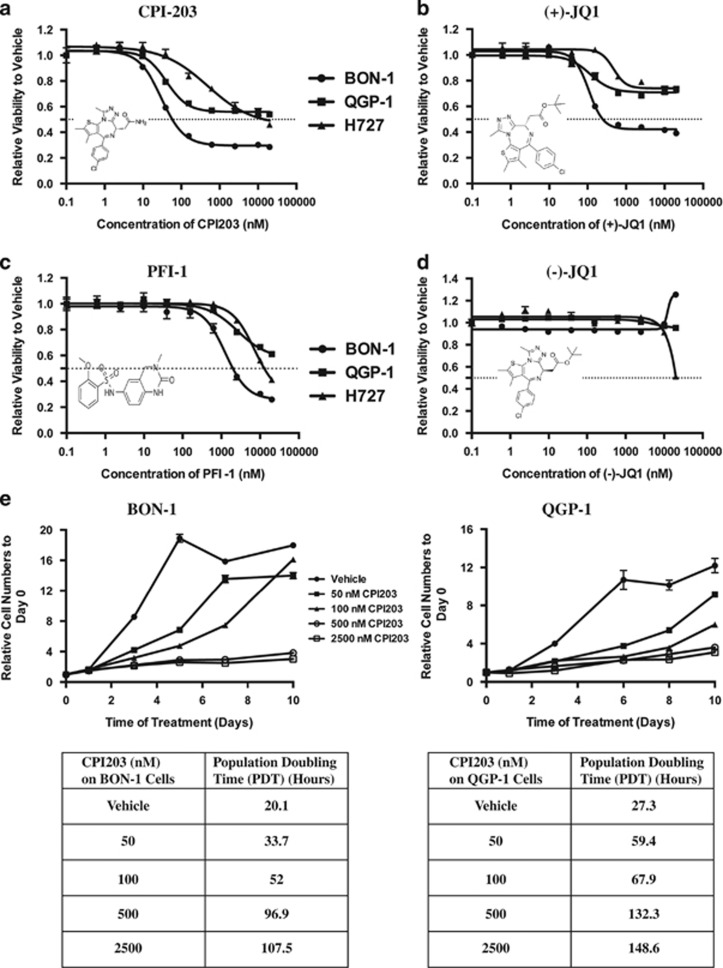
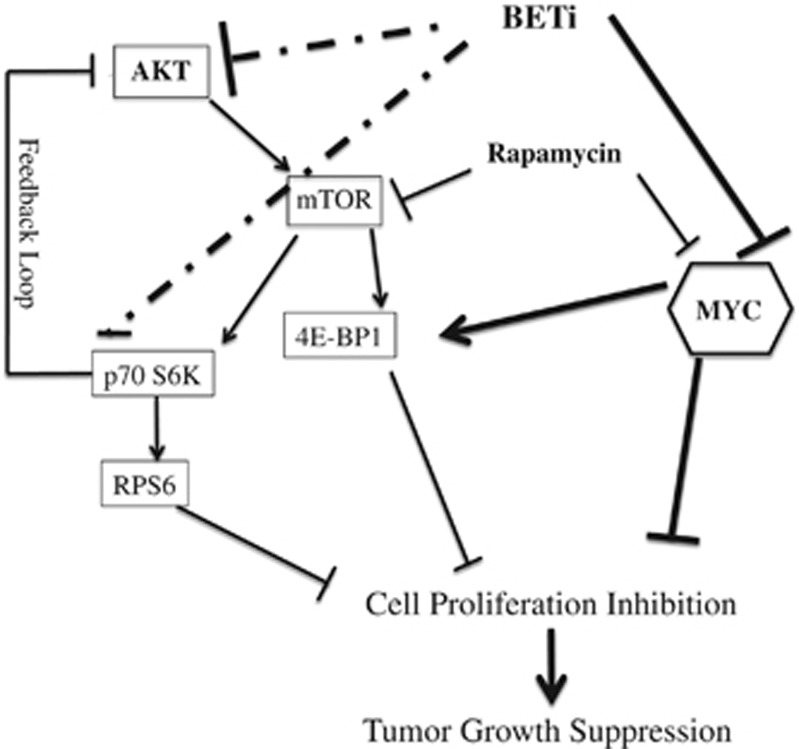
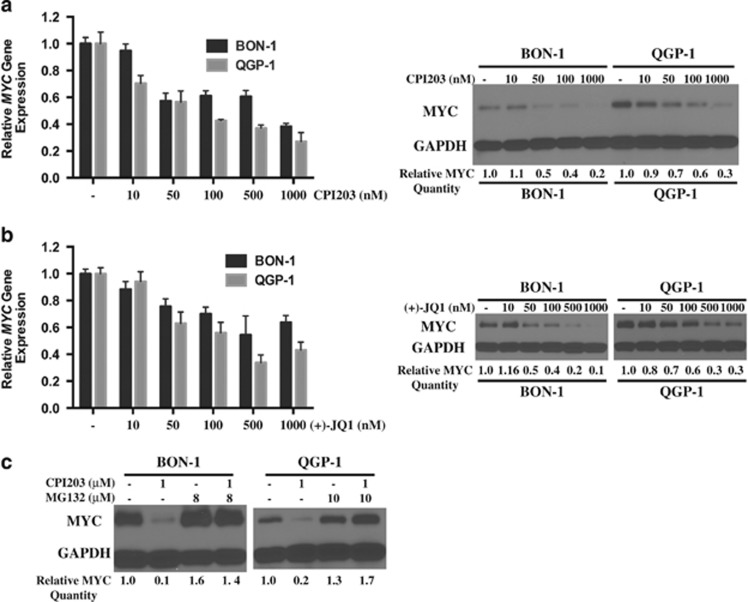
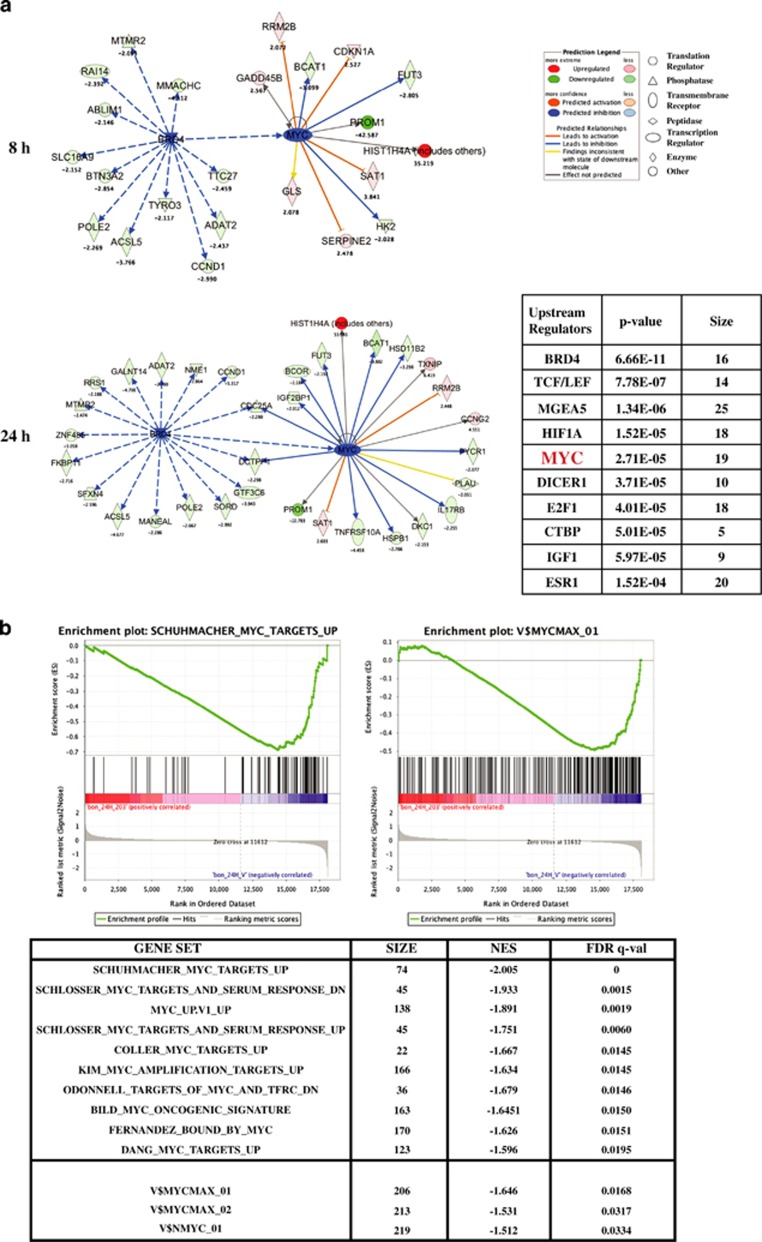
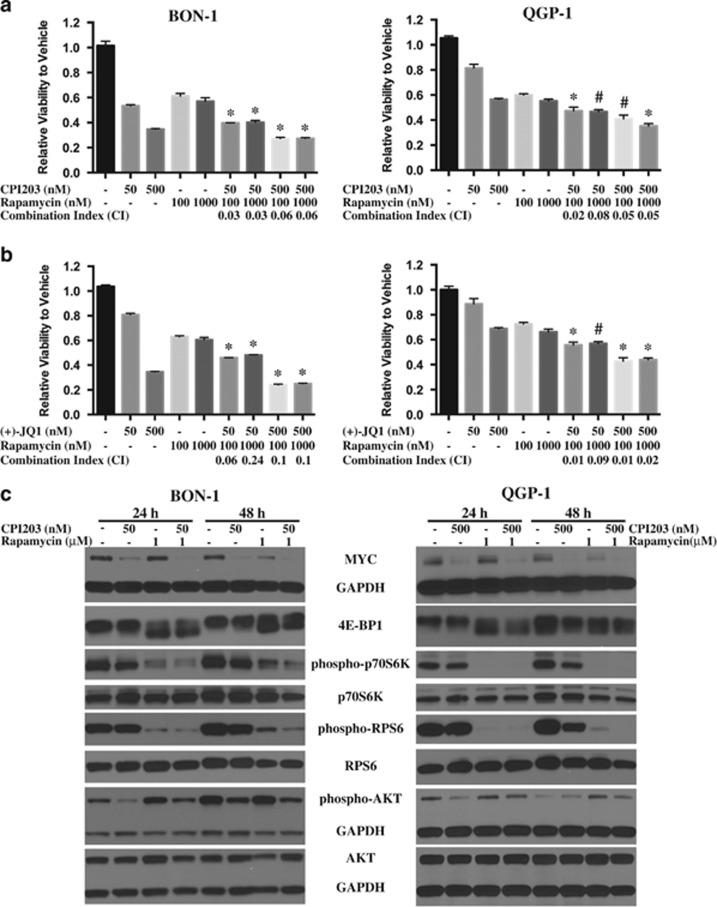
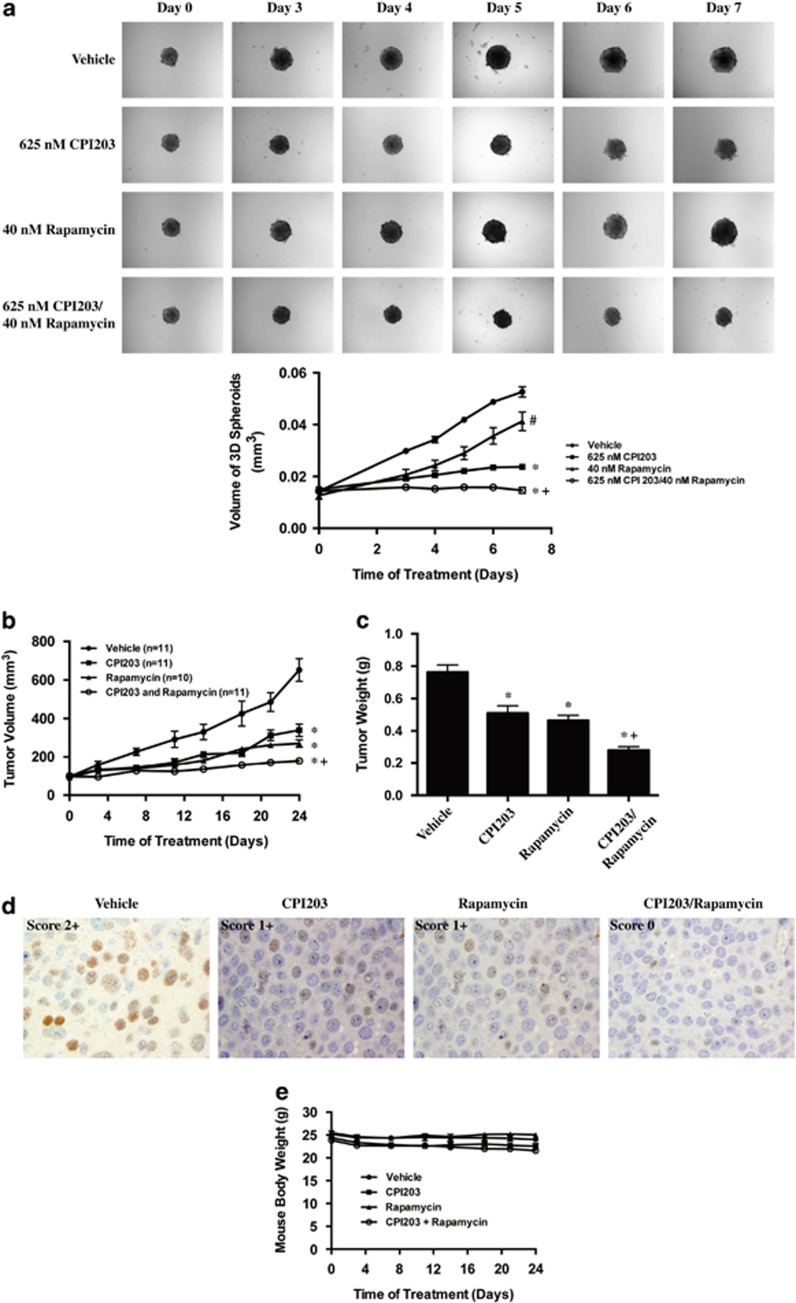
Endogenous c-MYC (MYC) has been reported to be a potential pharmacological target to trigger ubiquitous tumor regression of pancreatic neuroendocrine tumors (PanNETs) and lung tumors. Recently inhibitors of bromodomain and extra-terminal (BET) family proteins have shown antitumor effects through the suppression of MYC in leukemia and lymphoma. In this paper, we investigated the antitumor activity of a BET protein bromodomain inhibitor (BETi) CPI203 as a single agent and in combination with rapamycin in human PanNETs. We found that exposure of human PanNET cell lines to CPI203 led to downregulation of MYC expression, G1 cell cycle arrest and nearly complete inhibition of cell proliferation. In addition, overexpression of MYC suppressed the growth inhibition caused by CPI203 and knockdown of MYC phenocopied the effects of CPI203 treatment. These findings indicate that suppression of MYC contributed to the antiproliferative effects of BETi inhibition in human PanNET cells. Importantly, CPI203 treatment enhanced the antitumor effects of rapamycin in PanNET cells grown in monolayer and in three-dimensional cell cultures, as well as in a human PanNET xenograft model in vivo. Furthermore, the combination treatment attenuated rapamycin-induced AKT activation, a major limitation of rapamycin therapy. Collectively, our data suggest that targeting MYC with a BETi may increase the therapeutic benefits of rapalogs in human PanNET patients. This provides a novel clinical strategy for PanNETs, and possibly for other tumors as well.
据报道,内源性c-MYC(MYC)是触发胰腺神经内分泌肿瘤(PanNETs)和肺部肿瘤普遍肿瘤消退的潜在药理学靶点。最近,溴结构域和额外末端(BET)家族蛋白抑制剂已通过抑制白血病和淋巴瘤中的MYC显示出抗肿瘤作用。在本文中,我们研究了BET蛋白溴结构域抑制剂(BETi)CPI203作为单一药物以及与雷帕霉素联合在人PanNETs中的抗肿瘤活性。我们发现,将人PanNET细胞系暴露于CPI203会导致MYC表达下调、G1期细胞周期停滞以及细胞增殖几乎完全受到抑制。此外,MYC的过表达抑制了CPI203引起的生长抑制,而敲低MYC则模拟了CPI203处理的效果。这些发现表明,抑制MYC有助于BETi抑制在人PanNET细胞中的抗增殖作用。重要的是,CPI203处理增强了雷帕霉素在单层培养和三维细胞培养的PanNET细胞以及体内人PanNET异种移植模型中的抗肿瘤作用。此外,联合治疗减弱了雷帕霉素诱导的AKT激活,这是雷帕霉素治疗的一个主要局限性。总体而言,我们的数据表明,用BETi靶向MYC可能会增加雷帕霉素类似物对人PanNET患者的治疗益处。这为PanNETs提供了一种新的临床策略,也可能为其他肿瘤提供策略。