Authors' Affiliations: Department of Molecular Pharmacology and Therapeutics, Oncology Research Institute, Loyola University Chicago, Stritch School of Medicine, Maywood, Illinois; Departments of Medical Oncology and Radiation Oncology; Belfer Institute for Applied Cancer Science; Ludwig Center at Dana-Farber/Harvard Cancer Center; Lowe Center for Thoracic Oncology, Dana-Farber Cancer Institute; Department of Medicine, Brigham and Women's Hospital; Department of Medicine, Harvard Medical School, Boston, Massachusetts; Departament de Fisiologia, Facultat de Farmàcia, Universitat de València, Valencia, Spain; and Department of Pediatrics, Columbia University Medical Center, New York, New York.
Clin Cancer Res. 2013 Nov 15;19(22):6183-92. doi: 10.1158/1078-0432.CCR-12-3904. Epub 2013 Sep 17.
Amplification of MYC is one of the most common genetic alterations in lung cancer, contributing to a myriad of phenotypes associated with growth, invasion, and drug resistance. Murine genetics has established both the centrality of somatic alterations of Kras in lung cancer, as well as the dependency of mutant Kras tumors on MYC function. Unfortunately, drug-like small-molecule inhibitors of KRAS and MYC have yet to be realized. The recent discovery, in hematologic malignancies, that bromodomain and extra-terminal (BET) bromodomain inhibition impairs MYC expression and MYC transcriptional function established the rationale of targeting KRAS-driven non-small cell lung cancer (NSCLC) with BET inhibition.
We performed functional assays to evaluate the effects of JQ1 in genetically defined NSCLC cell lines harboring KRAS and/or LKB1 mutations. Furthermore, we evaluated JQ1 in transgenic mouse lung cancer models expressing mutant kras or concurrent mutant kras and lkb1. Effects of bromodomain inhibition on transcriptional pathways were explored and validated by expression analysis.
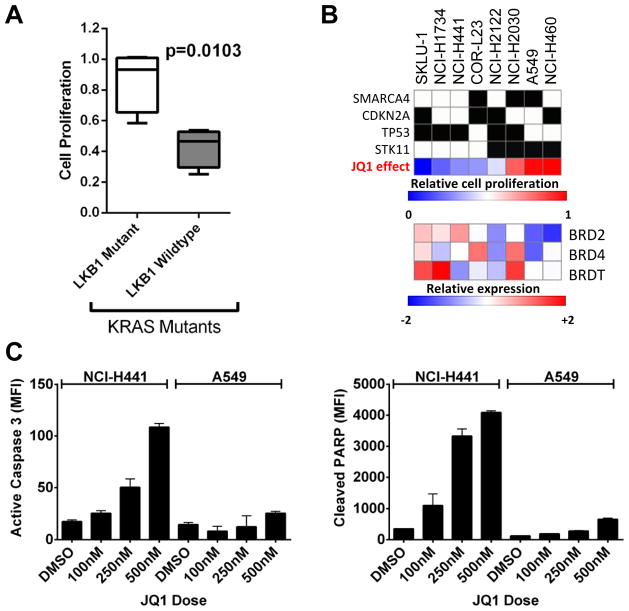
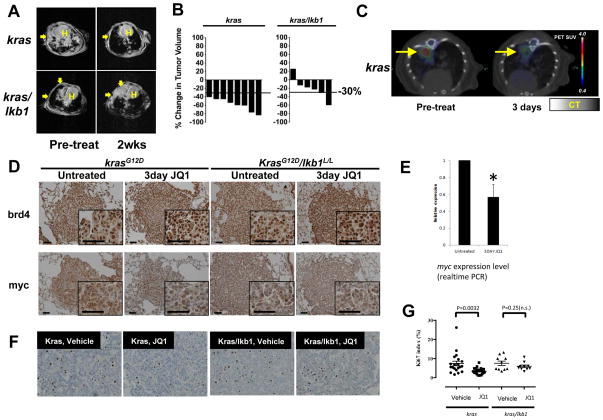
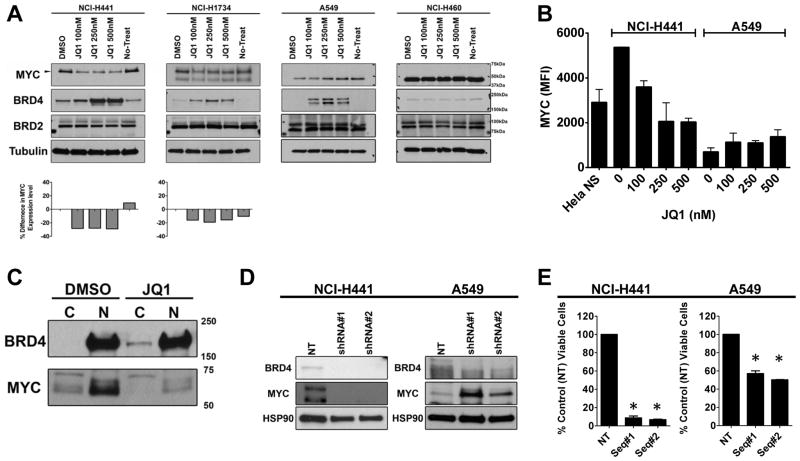
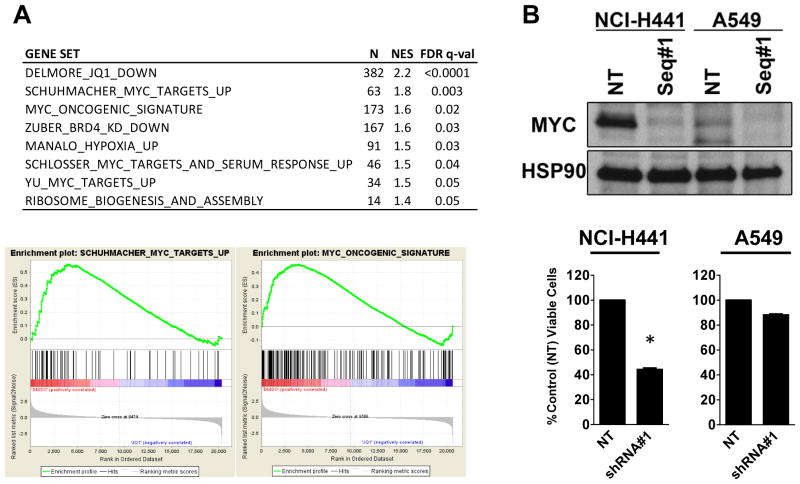
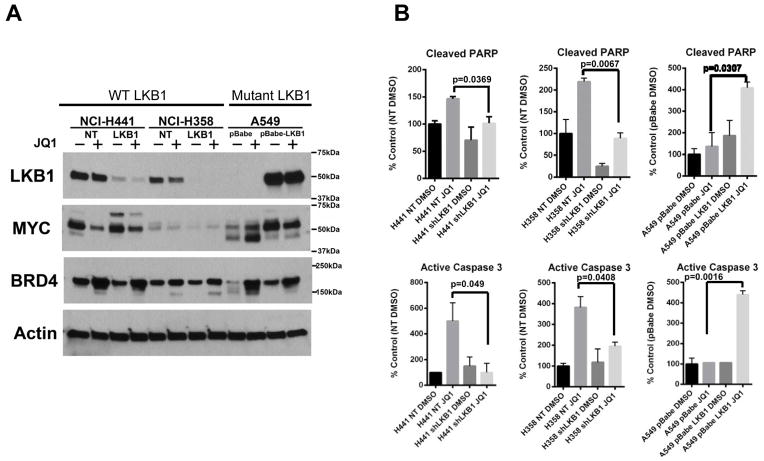
Although JQ1 is broadly active in NSCLC cells, activity of JQ1 in mutant KRAS NSCLC is abrogated by concurrent alteration or genetic knockdown of LKB1. In sensitive NSCLC models, JQ1 treatment results in the coordinate downregulation of the MYC-dependent transcriptional program. We found that JQ1 treatment produces significant tumor regression in mutant kras mice. As predicted, tumors from mutant kras and lkb1 mice did not respond to JQ1.
Bromodomain inhibition comprises a promising therapeutic strategy for KRAS-mutant NSCLC with wild-type LKB1, via inhibition of MYC function. Clinical studies of BET bromodomain inhibitors in aggressive NSCLC will be actively pursued. Clin Cancer Res; 19(22); 6183-92. ©2013 AACR.
MYC 扩增是肺癌中最常见的遗传改变之一,导致与生长、侵袭和耐药性相关的多种表型。鼠遗传学已经确立了 Kras 体细胞改变在肺癌中的核心地位,以及突变 Kras 肿瘤对 MYC 功能的依赖性。不幸的是,KRAS 和 MYC 的药物样小分子抑制剂尚未实现。最近在血液恶性肿瘤中发现,溴结构域和末端(BET)溴结构域抑制会损害 MYC 表达和 MYC 转录功能,这为用 BET 抑制靶向 KRAS 驱动的非小细胞肺癌(NSCLC)提供了依据。
我们进行了功能测定,以评估 JQ1 在携带 KRAS 和/或 LKB1 突变的遗传定义的 NSCLC 细胞系中的作用。此外,我们在表达突变 kras 或同时表达突变 kras 和 lkb1 的转基因小鼠肺癌模型中评估了 JQ1。通过表达分析探讨和验证了溴结构域抑制对转录途径的影响。
尽管 JQ1 在 NSCLC 细胞中广泛活跃,但 KRAS NSCLC 中 JQ1 的活性被 LKB1 的并发改变或基因敲低所阻断。在敏感的 NSCLC 模型中,JQ1 治疗导致 MYC 依赖性转录程序的协调下调。我们发现 JQ1 治疗可使突变 kras 小鼠的肿瘤明显消退。正如所预测的那样,突变 kras 和 lkb1 小鼠的肿瘤对 JQ1 没有反应。
通过抑制 MYC 功能,溴结构域抑制构成了具有野生型 LKB1 的 KRAS 突变型 NSCLC 的一种很有前途的治疗策略。将积极开展 BET 溴结构域抑制剂在侵袭性 NSCLC 中的临床研究。临床癌症研究;19(22);6183-92。©2013AACR。