Chowdhury Sandipan, Haehnel Benjamin M, Chanda Baron
Graduate Program in Biophysics and Department of Neuroscience, University of Wisconsin, Madison, WI 53705 Graduate Program in Biophysics and Department of Neuroscience, University of Wisconsin, Madison, WI 53705.
Graduate Program in Biophysics and Department of Neuroscience, University of Wisconsin, Madison, WI 53705 Graduate Program in Biophysics and Department of Neuroscience, University of Wisconsin, Madison, WI 53705
J Gen Physiol. 2014 Nov;144(5):441-55. doi: 10.1085/jgp.201411184. Epub 2014 Oct 13.
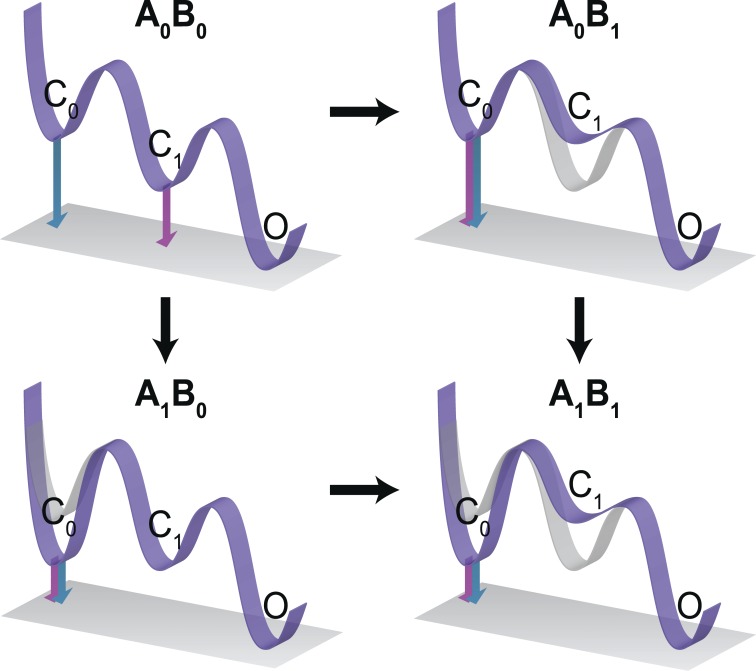
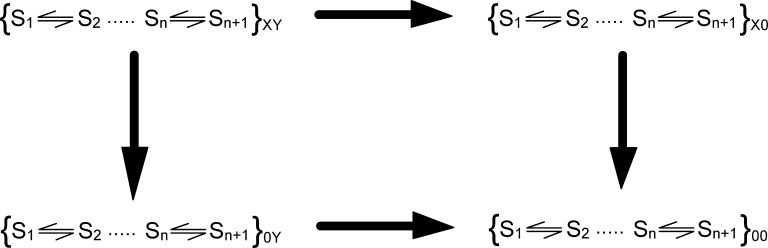
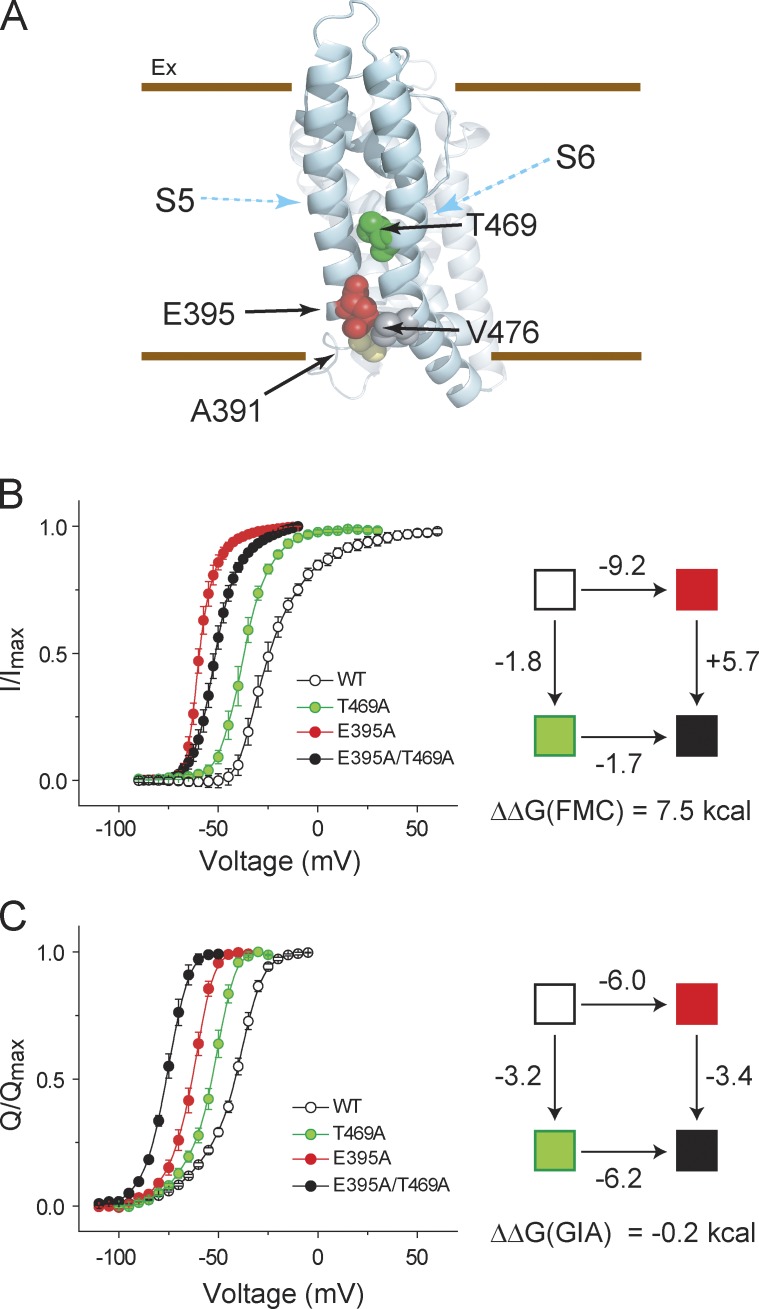
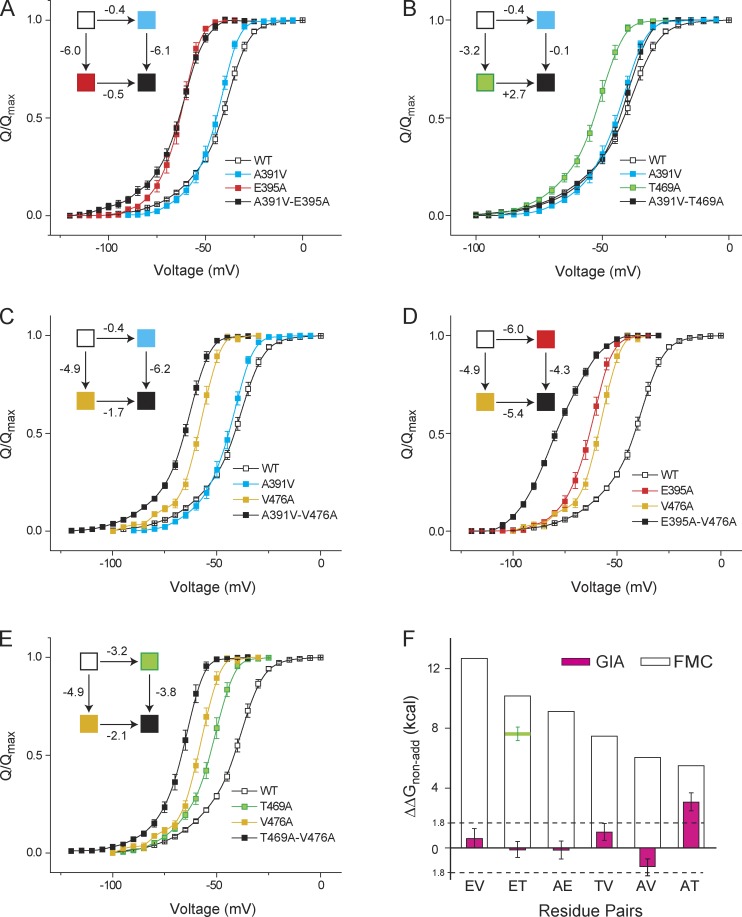
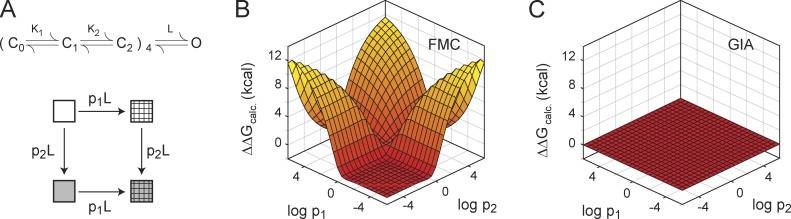
Signaling proteins such as ion channels largely exist in two functional forms, corresponding to the active and resting states, connected by multiple intermediates. Multiparametric kinetic models based on sophisticated electrophysiological experiments have been devised to identify molecular interactions of these conformational transitions. However, this approach is arduous and is not suitable for large-scale perturbation analysis of interaction pathways. Recently, we described a model-free method to obtain the net free energy of activation in voltage- and ligand-activated ion channels. Here we extend this approach to estimate pairwise interaction energies of side chains that contribute to gating transitions. Our approach, which we call generalized interaction-energy analysis (GIA), combines median voltage estimates obtained from charge-voltage curves with mutant cycle analysis to ascertain the strengths of pairwise interactions. We show that, for a system with an arbitrary gating scheme, the nonadditive contributions of amino acid pairs to the net free energy of activation can be computed in a self-consistent manner. Numerical analyses of sequential and allosteric models of channel activation also show that this approach can measure energetic nonadditivities even when perturbations affect multiple transitions. To demonstrate the experimental application of this method, we reevaluated the interaction energies of six previously described long-range interactors in the Shaker potassium channel. Our approach offers the ability to generate detailed interaction energy maps in voltage- and ligand-activated ion channels and can be extended to any force-driven system as long as associated "displacement" can be measured.
诸如离子通道等信号蛋白大多以两种功能形式存在,分别对应于活性状态和静息状态,二者由多个中间状态相连。基于精密电生理实验设计的多参数动力学模型已被用于识别这些构象转变的分子相互作用。然而,这种方法很艰巨,不适用于对相互作用途径进行大规模扰动分析。最近,我们描述了一种无模型方法来获取电压门控和配体门控离子通道中的净活化自由能。在此,我们扩展这种方法来估计有助于门控转变的侧链间的成对相互作用能。我们的方法,即广义相互作用能分析(GIA),将从电荷 - 电压曲线获得的中值电压估计与突变循环分析相结合,以确定成对相互作用的强度。我们表明,对于具有任意门控机制的系统,氨基酸对净活化自由能的非加和贡献可以以自洽的方式计算。通道激活的顺序模型和别构模型的数值分析也表明,即使扰动影响多个转变,这种方法也能测量能量非加和性。为了证明该方法的实验应用,我们重新评估了先前描述的摇椅式钾通道中六个长程相互作用因子的相互作用能。我们的方法能够在电压门控和配体门控离子通道中生成详细的相互作用能图谱,并且只要能够测量相关的“位移”,就可以扩展到任何力驱动系统。