Patiño Liliana Catherine, Battu Rajani, Ortega-Recalde Oscar, Nallathambi Jeyabalan, Anandula Venkata Ramana, Renukaradhya Umashankar, Laissue Paul
Unidad de Genética, Grupo GENIUROS, Escuela de Medicina y Ciencias de la Salud, Universidad del Rosario, Bogotá, Colombia.
Department of Vitreoretina, Narayana Nethralaya, Bangalore, India.
PLoS One. 2014 Oct 15;9(10):e109576. doi: 10.1371/journal.pone.0109576. eCollection 2014.
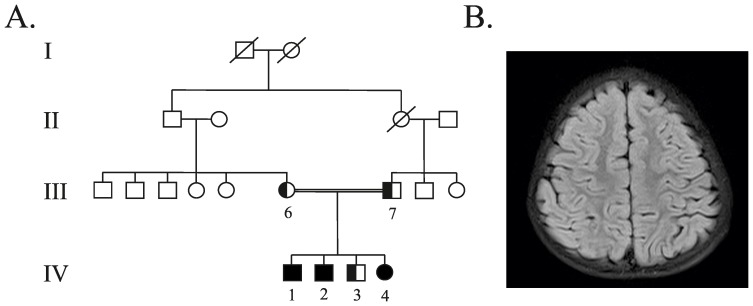
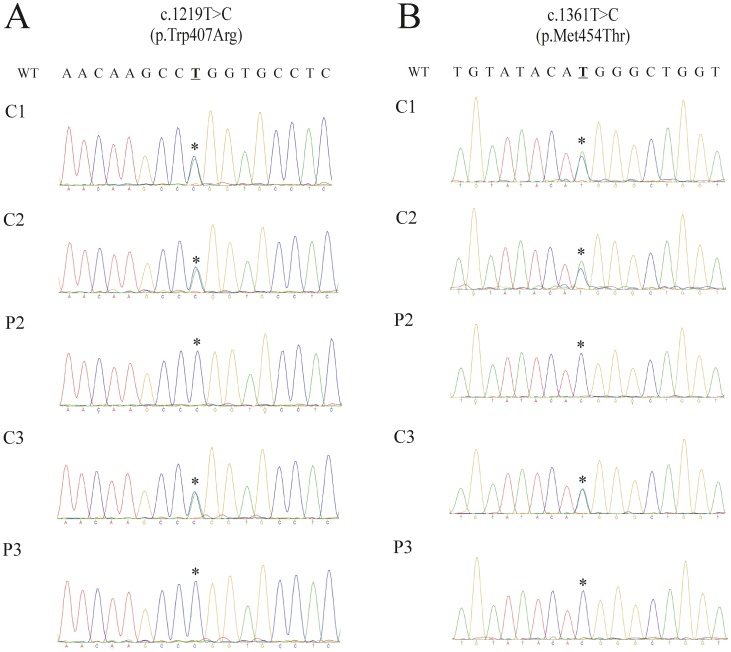
The neuronal ceroid-lipofuscinoses (NCL) is a group of neurodegenerative disorders characterized by epilepsy, visual failure, progressive mental and motor deterioration, myoclonus, dementia and reduced life expectancy. Classically, NCL-affected individuals have been classified into six categories, which have been mainly defined regarding the clinical onset of symptoms. However, some patients cannot be easily included in a specific group because of significant variation in the age of onset and disease progression. Molecular genetics has emerged in recent years as a useful tool for enhancing NCL subtype classification. Fourteen NCL genetic forms (CLN1 to CLN14) have been described to date. The variant late-infantile form of the disease has been linked to CLN5, CLN6, CLN7 (MFSD8) and CLN8 mutations. Despite advances in the diagnosis of neurodegenerative disorders mutations in these genes may cause similar phenotypes, which rends difficult accurate candidate gene selection for direct sequencing. Three siblings who were affected by variant late-infantile NCL are reported in the present study. We used whole-exome sequencing, direct sequencing and in silico approaches to identify the molecular basis of the disease. We identified the novel c.1219T>C (p.Trp407Arg) and c.1361T>C (p.Met454Thr) MFSD8 pathogenic mutations. Our results highlighted next generation sequencing as a novel and powerful methodological approach for the rapid determination of the molecular diagnosis of NCL. They also provide information regarding the phenotypic and molecular spectrum of CLN7 disease.
神经元蜡样脂褐质沉积症(NCL)是一组神经退行性疾病,其特征为癫痫、视力减退、进行性精神和运动功能衰退、肌阵挛、痴呆以及预期寿命缩短。传统上,受NCL影响的个体被分为六类,主要是根据症状的临床发作情况来定义的。然而,由于发病年龄和疾病进展存在显著差异,一些患者难以轻易归入某一特定类别。近年来,分子遗传学已成为增强NCL亚型分类的有用工具。迄今为止,已描述了14种NCL基因形式(CLN1至CLN14)。该疾病的变异型晚发性婴儿型已与CLN5、CLN6、CLN7(MFSD8)和CLN8突变相关。尽管神经退行性疾病的诊断取得了进展,但这些基因中的突变可能导致相似的表型,这使得难以准确选择直接测序的候选基因。本研究报告了三名受变异型晚发性婴儿型NCL影响的兄弟姐妹。我们使用全外显子测序、直接测序和计算机分析方法来确定该疾病的分子基础。我们鉴定出了新的c.1219T>C(p.Trp407Arg)和c.1361T>C(p.Met454Thr)MFSD8致病突变。我们的结果突出了下一代测序作为一种新颖且强大的方法,可快速确定NCL的分子诊断。它们还提供了有关CLN7疾病的表型和分子谱的信息。