Department of Neurology, Third Xiangya Hospital, Central South University, Changsha 410013, Hunan Province, China.
Department of Ultrastructure, School of Basic Medical Science, Central South University, Changsha 410078, Hunan Province, China.
Neural Regen Res. 2012 Nov 15;7(32):2522-7. doi: 10.3969/j.issn.1673-5374.2012.32.006.
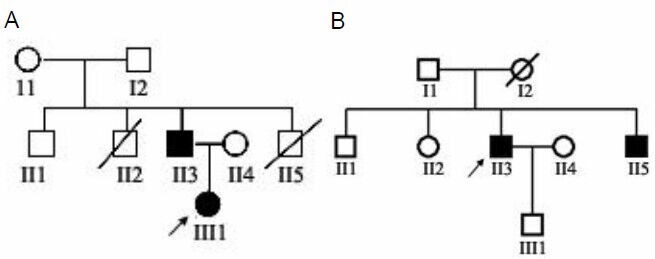
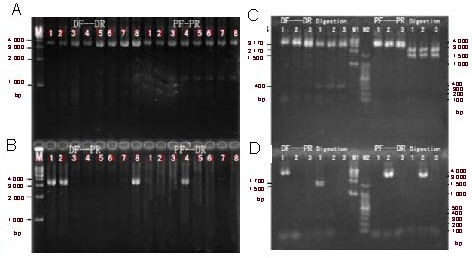
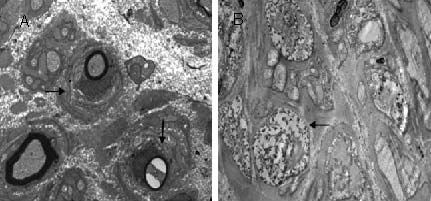
We used the allele-specific PCR-double digestion method on peripheral myelin protein 22 (PMP22) to determine duplication and deletion mutations in the proband and family members of one family with Charcot-Marie-Tooth disease type 1 and one family with hereditary neuropathy with liability to pressure palsies. The proband and one subclinical family member from the Charcot-Marie-Tooth disease type 1 family had a PMP22 gene duplication; one patient from the hereditary neuropathy with liability to pressure palsies family had a PMP22 gene deletion. Electron microscopic analysis of ultrathin sections of the superficial peroneal nerve from the two probands demonstrated demyelination and myelin sheath hyperplasia, as well as an 'onion-like' structure in the Charcot-Marie-Tooth disease type 1A patient. We observed an irregular thickened myelin sheath and 'mouse-nibbled'-like changes in the patient with hereditary neuropathy with liability to pressure palsies. In the Charcot-Marie-Tooth disease type 1A patient, nerve electrophysiological examination revealed moderate-to-severe reductions in the motor and sensory conduction velocities of the bilateral median nerve, ulnar nerve, tibial nerve, and sural nerve. Moreover, the compound muscle action potential amplitude was decreased. In the patient with hereditary neuropathy with liability to pressure palsies, the nerve conduction velocity of the bilateral tibial nerve and sural nerve was moderately reduced, and the nerve conduction velocity of the median nerve and ulnar nerve of both upper extremities was slightly reduced.
我们使用外周髓鞘蛋白 22(PMP22)的等位基因特异性 PCR-双重消化方法,在 1 个遗传性运动感觉神经病和 1 个遗传性压力易感性神经病家系的先证者及其家系成员中检测到 PMP22 基因的重复和缺失突变。1 例遗传性运动感觉神经病家系的先证者和 1 例亚临床家系成员存在 PMP22 基因重复;1 例遗传性压力易感性神经病家系的患者存在 PMP22 基因缺失。2 例先证者腓浅神经超微结构分析显示脱髓鞘和髓鞘增生,以及 1A 型遗传性运动感觉神经病患者的“洋葱样”结构。我们观察到遗传性压力易感性神经病患者的不规则增厚的髓鞘和“鼠噬样”改变。在遗传性运动感觉神经病 1A 型患者中,神经电生理检查显示双侧正中神经、尺神经、胫神经和腓总神经的运动和感觉传导速度均有中度至重度降低。此外,复合肌肉动作电位幅度降低。在遗传性压力易感性神经病患者中,双侧胫神经和腓总神经的神经传导速度中度降低,双侧上肢正中神经和尺神经的神经传导速度轻度降低。