Eppinger Mark, Pearson Talima, Koenig Sara S K, Pearson Ofori, Hicks Nathan, Agrawal Sonia, Sanjar Fatemeh, Galens Kevin, Daugherty Sean, Crabtree Jonathan, Hendriksen Rene S, Price Lance B, Upadhyay Bishnu P, Shakya Geeta, Fraser Claire M, Ravel Jacques, Keim Paul S
Center for Microbial Genetics and Genomics, Northern Arizona University, Flagstaff, Arizona, USA.
Department of Biology, University of Texas, San Antonio, Texas, USA.
mBio. 2014 Nov 4;5(6):e01721. doi: 10.1128/mBio.01721-14.
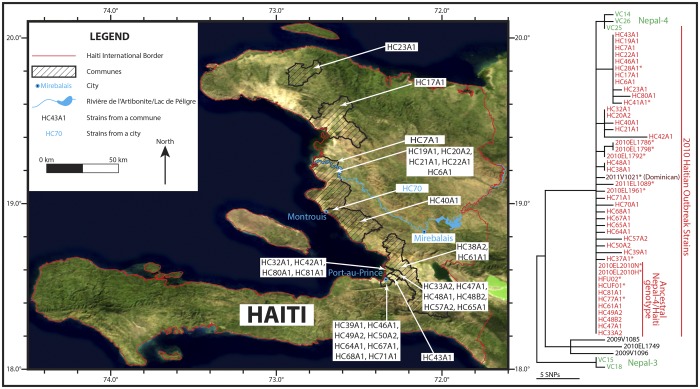
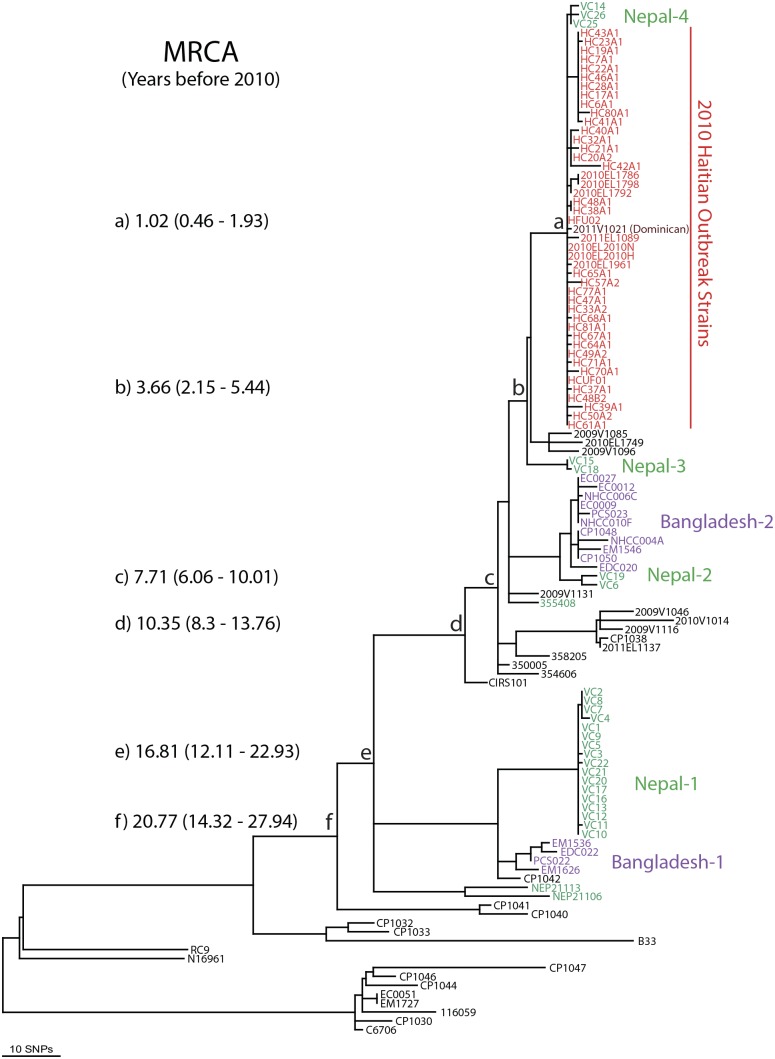
For centuries, cholera has been one of the most feared diseases. The causative agent Vibrio cholerae is a waterborne Gram-negative enteric pathogen eliciting a severe watery diarrheal disease. In October 2010, the seventh pandemic reached Haiti, a country that had not experienced cholera for more than a century. By using whole-genome sequence typing and mapping strategies of 116 serotype O1 strains from global sources, including 44 Haitian genomes, we present a detailed reconstructed evolutionary history of the seventh pandemic with a focus on the Haitian outbreak. We catalogued subtle genomic alterations at the nucleotide level in the genome core and architectural rearrangements from whole-genome map comparisons. Isolates closely related to the Haitian isolates caused several recent outbreaks in southern Asia. This study provides evidence for a single-source introduction of cholera from Nepal into Haiti followed by rapid, extensive, and continued clonal expansion. The phylogeographic patterns in both southern Asia and Haiti argue for the rapid dissemination of V. cholerae across the landscape necessitating real-time surveillance efforts to complement the whole-genome epidemiological analysis. As eradication efforts move forward, phylogeographic knowledge will be important for identifying persistent sources and monitoring success at regional levels. The results of molecular and epidemiological analyses of this outbreak suggest that an indigenous Haitian source of V. cholerae is unlikely and that an indigenous source has not contributed to the genomic evolution of this clade.
In this genomic epidemiology study, we have applied high-resolution whole-genome-based sequence typing methodologies on a comprehensive set of genome sequences that have become available in the aftermath of the Haitian cholera epidemic. These sequence resources enabled us to reassess the degree of genomic heterogeneity within the Vibrio cholerae O1 serotype and to refine boundaries and evolutionary relationships. The established phylogenomic framework showed how outbreak isolates fit into the global phylogeographic patterns compared to a comprehensive globally and temporally diverse strain collection and provides strong molecular evidence that points to a nonindigenous source of the 2010 Haitian cholera outbreak and refines epidemiological standards used in outbreak investigations for outbreak inclusion/exclusion following the concept of genomic epidemiology. The generated phylogenomic data have major public health relevance in translating sequence-based information to assist in future diagnostic, epidemiological, surveillance, and forensic studies of cholera.
几个世纪以来,霍乱一直是最令人恐惧的疾病之一。病原体霍乱弧菌是一种通过水传播的革兰氏阴性肠道病原体,可引发严重的水样腹泻疾病。2010年10月,第七次霍乱大流行蔓延至海地,这个国家已有一个多世纪未曾经历过霍乱疫情。通过对来自全球的116株O1血清型菌株(包括44株海地菌株)进行全基因组序列分型和图谱绘制策略,我们呈现了第七次霍乱大流行详细的重建进化史,重点关注海地疫情爆发情况。我们梳理了基因组核心区域核苷酸水平上的细微基因组改变以及全基因组图谱比较中的结构重排。与海地分离株密切相关的菌株在南亚引发了几次近期疫情。本研究为霍乱从尼泊尔单源传入海地,随后进行快速、广泛且持续的克隆扩张提供了证据。南亚和海地的系统发育地理模式表明霍乱弧菌在各地迅速传播,这就需要进行实时监测,以补充全基因组流行病学分析。随着根除霍乱的努力推进,系统发育地理学知识对于识别持续的传染源以及监测区域层面的成效将至关重要。此次疫情的分子和流行病学分析结果表明,海地霍乱弧菌不太可能源自本土,且本土来源并未对该进化枝的基因组进化产生影响。
在这项基因组流行病学研究中,我们对海地霍乱疫情后可获取的一组全面的基因组序列应用了基于全基因组的高分辨率序列分型方法。这些序列资源使我们能够重新评估霍乱弧菌O1血清型内的基因组异质性程度,并完善界限和进化关系。所建立的系统发育基因组框架展示了与全球范围内全面且时空多样的菌株集合相比,疫情爆发分离株如何融入全球系统发育地理模式,并提供了有力的分子证据,表明2010年海地霍乱疫情的源头并非本土,并完善了疫情调查中用于根据基因组流行病学概念纳入/排除疫情的流行病学标准。所生成的系统发育基因组数据在将基于序列的信息转化以协助未来霍乱的诊断、流行病学、监测和法医研究方面具有重大公共卫生意义。