Ashkenazy Haim, Cohen Ofir, Pupko Tal, Huchon Dorothée
Department of Cell Research and Immunology, George S. Wise Faculty of Life Sciences, Tel Aviv University, Ramat Aviv, Israel.
Department of Cell Research and Immunology, George S. Wise Faculty of Life Sciences, Tel Aviv University, Ramat Aviv, Israel Present address: Department of Molecular Genetics, Weizmann Institute of Science, Rehovot, Israel.
Genome Biol Evol. 2014 Nov 18;6(12):3199-209. doi: 10.1093/gbe/evu252.
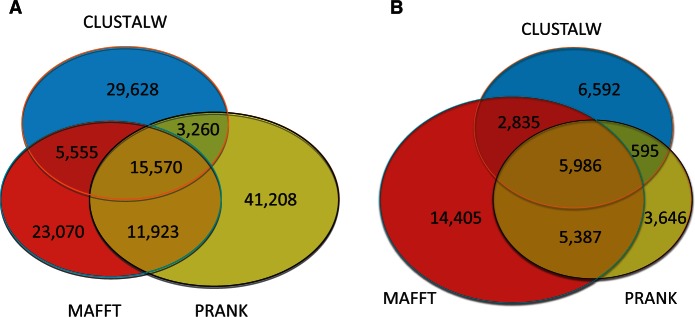
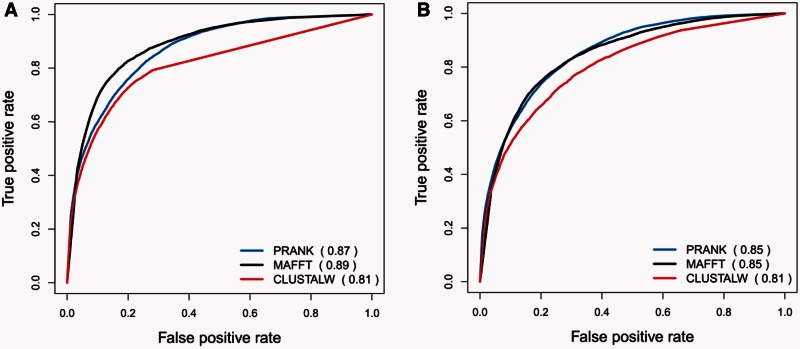
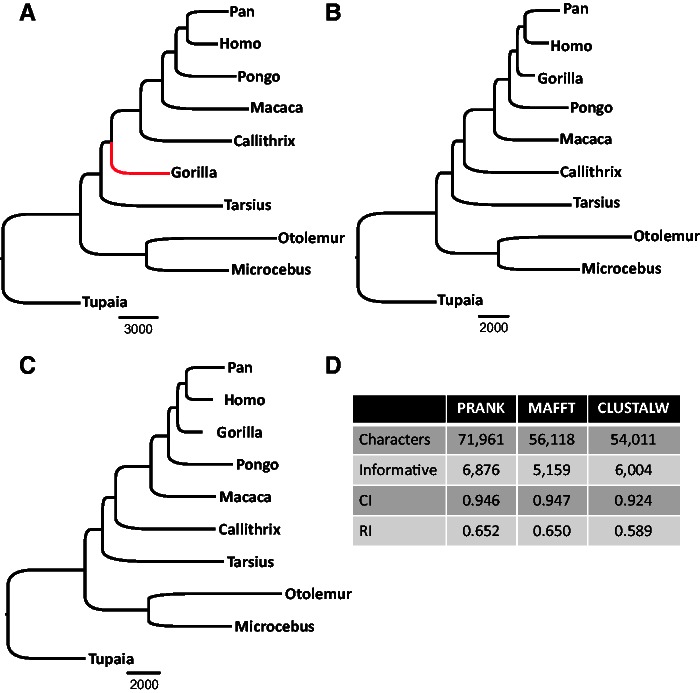
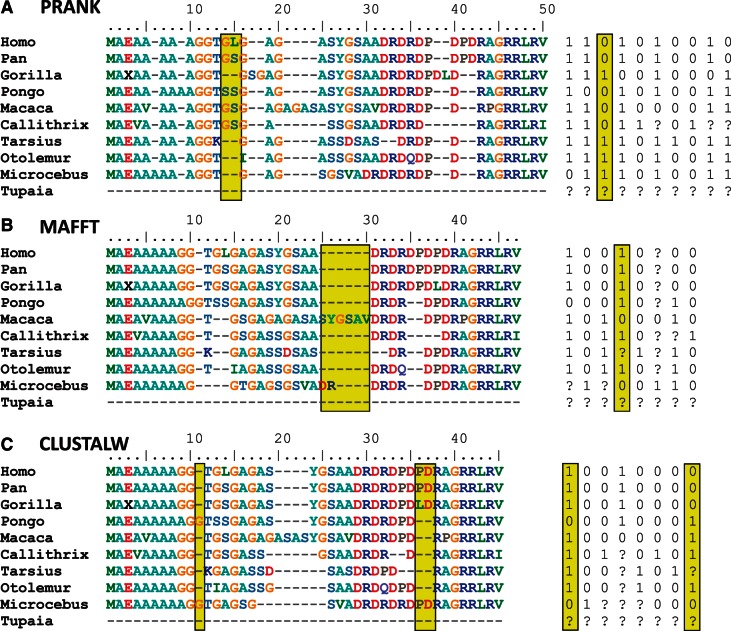
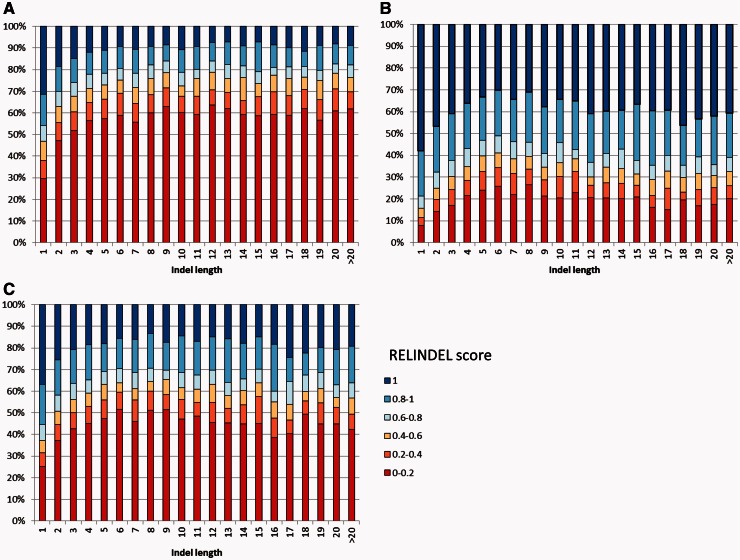
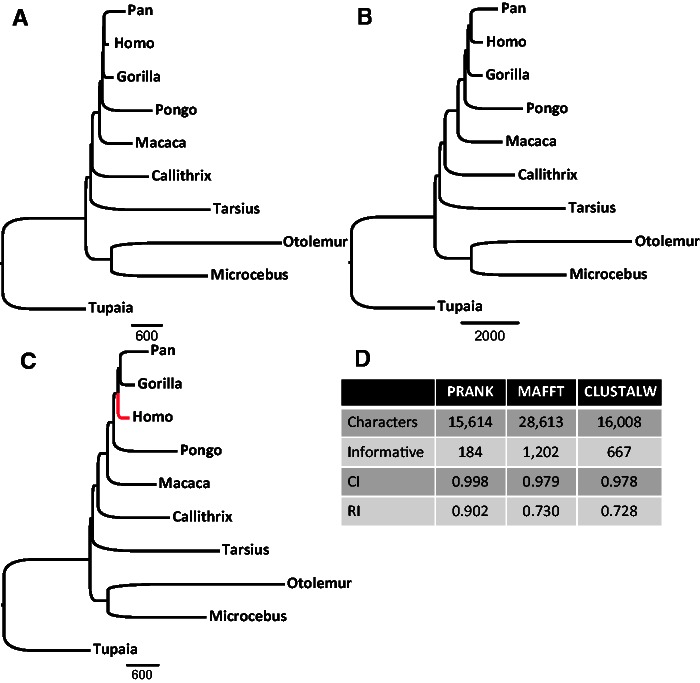
It is often assumed that it is unlikely that the same insertion or deletion (indel) event occurred at the same position in two independent evolutionary lineages, and thus, indel-based inference of phylogeny should be less subject to homoplasy compared with standard inference which is based on substitution events. Indeed, indels were successfully used to solve debated evolutionary relationships among various taxonomical groups. However, indels are never directly observed but rather inferred from the alignment and thus indel-based inference may be sensitive to alignment errors. It is hypothesized that phylogenetic reconstruction would be more accurate if it relied only on a subset of reliable indels instead of the entire indel data. Here, we developed a method to quantify the reliability of indel characters by measuring how often they appear in a set of alternative multiple sequence alignments. Our approach is based on the assumption that indels that are consistently present in most alternative alignments are more reliable compared with indels that appear only in a small subset of these alignments. Using simulated and empirical data, we studied the impact of filtering and weighting indels by their reliability scores on the accuracy of indel-based phylogenetic reconstruction. The new method is available as a web-server at http://guidance.tau.ac.il/RELINDEL/.
人们通常认为,在两个独立的进化谱系中,不太可能在同一位置发生相同的插入或缺失(indel)事件,因此,与基于替换事件的标准系统发育推断相比,基于indel的系统发育推断应该较少受到同塑性的影响。事实上,indel已成功用于解决不同分类群之间有争议的进化关系。然而,indel从未被直接观察到,而是从比对中推断出来的,因此基于indel的推断可能对齐列错误敏感。据推测,如果系统发育重建仅依赖于可靠indel的一个子集而不是整个indel数据,将会更准确。在这里,我们开发了一种方法,通过测量indel特征在一组替代多序列比对中出现的频率来量化其可靠性。我们的方法基于这样的假设:与仅出现在这些比对的一小部分中的indel相比,在大多数替代比对中一致出现的indel更可靠。使用模拟和实证数据,我们研究了根据indel的可靠性得分对其进行过滤和加权对基于indel的系统发育重建准确性的影响。新方法可作为网络服务器在http://guidance.tau.ac.il/RELINDEL/上获取。