Sowd Gregory A, Mody Dviti, Eggold Joshua, Cortez David, Friedman Katherine L, Fanning Ellen
Department of Biological Sciences, Vanderbilt University, Nashville, Tennessee, United States of America.
Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee, United States of America.
PLoS Pathog. 2014 Dec 4;10(12):e1004536. doi: 10.1371/journal.ppat.1004536. eCollection 2014 Dec.
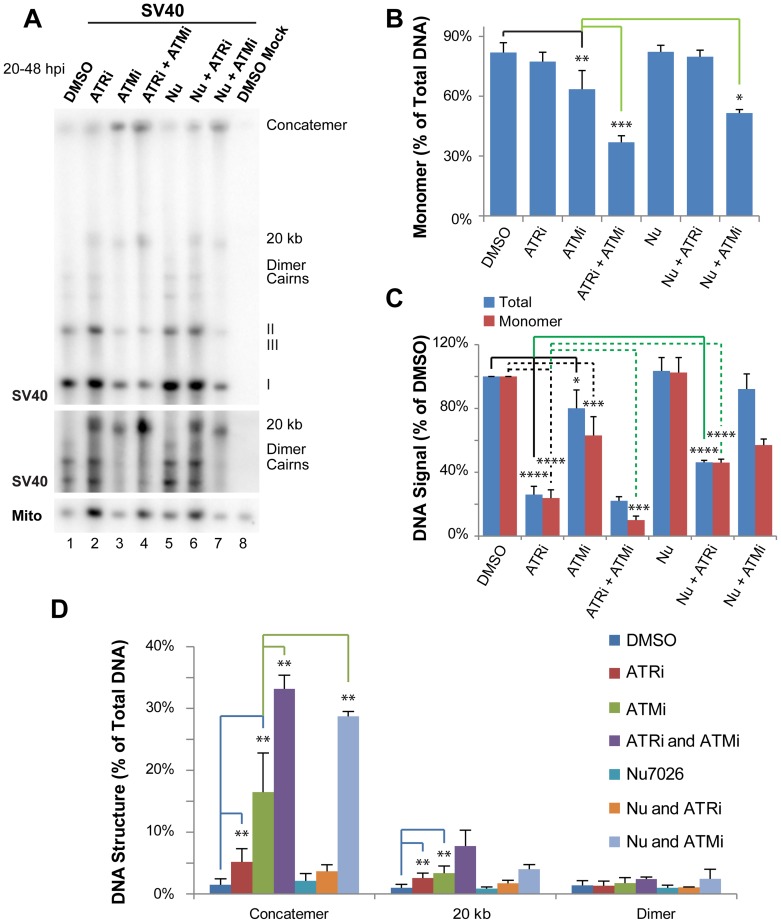
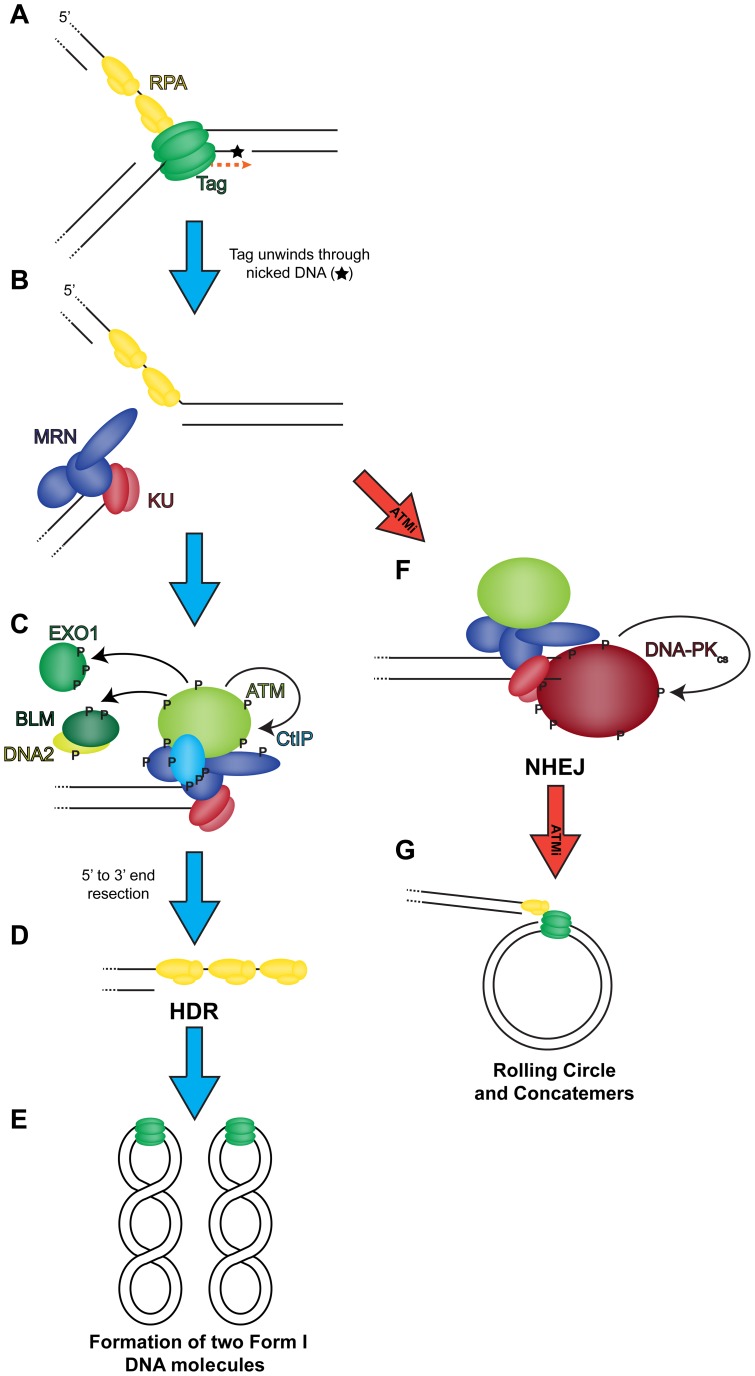
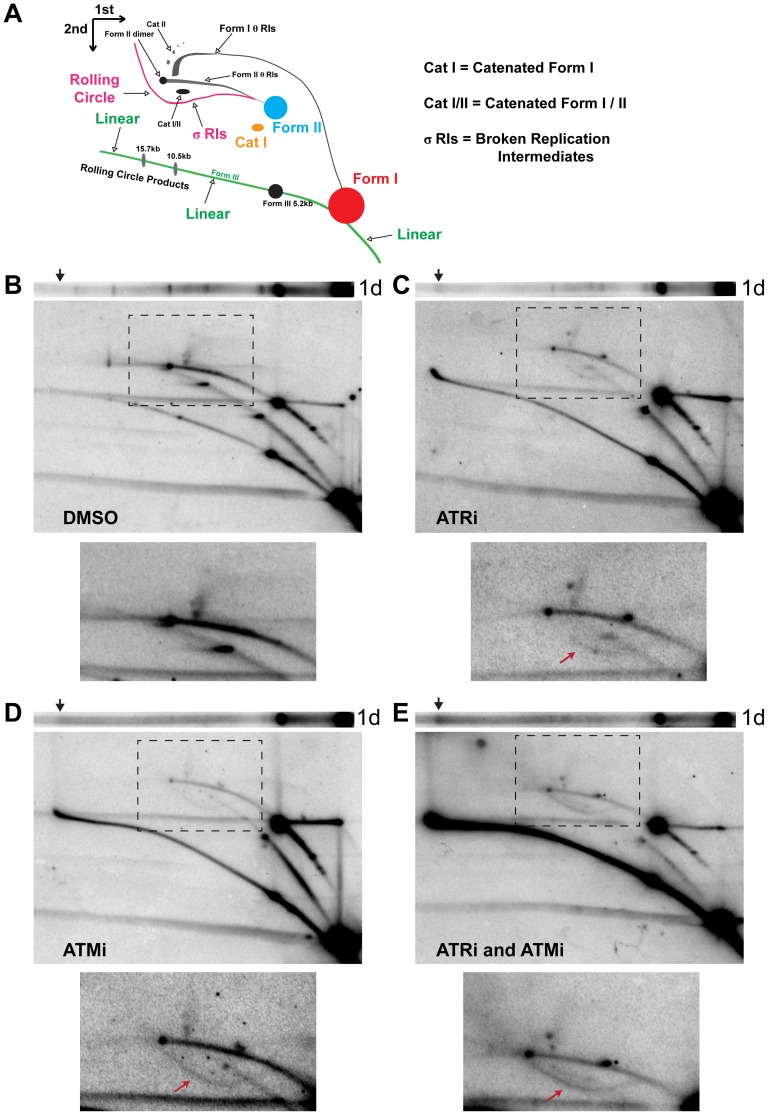
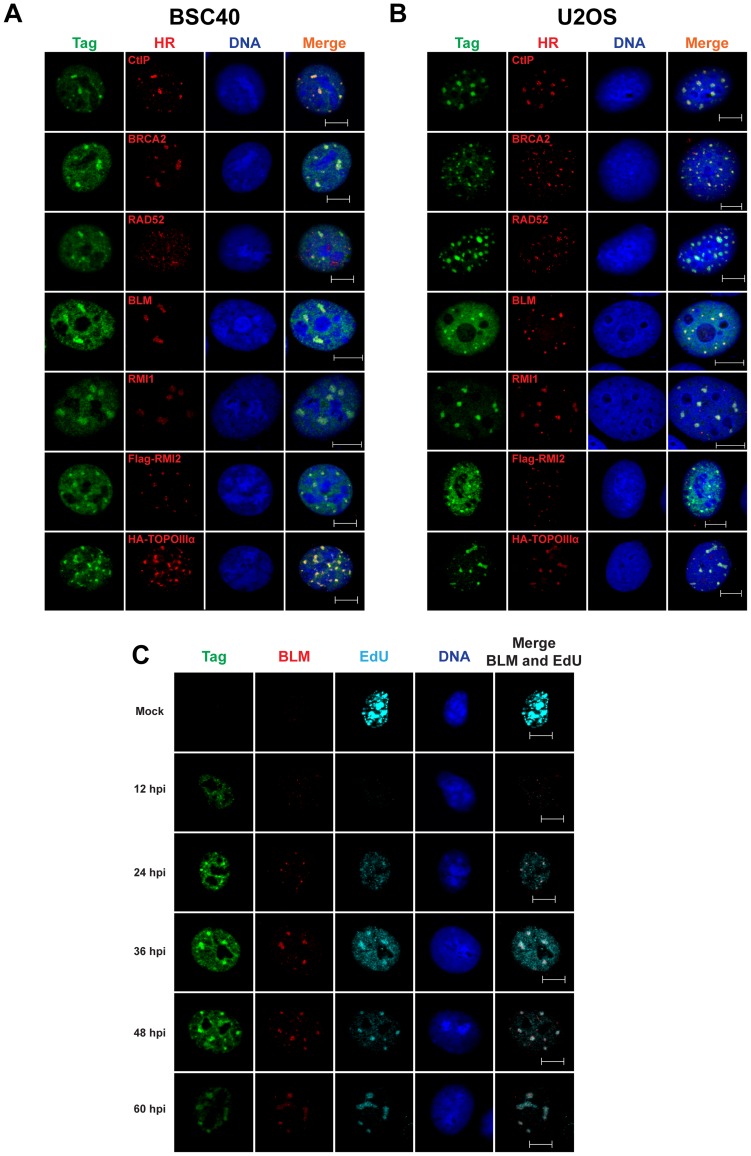
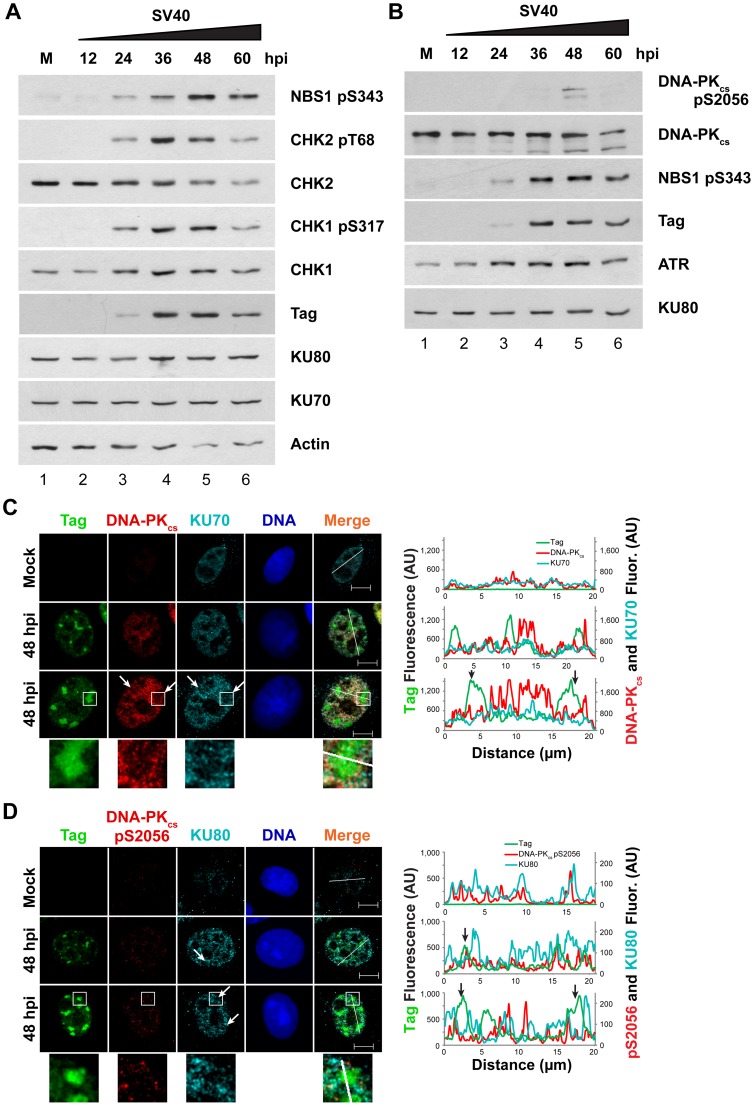
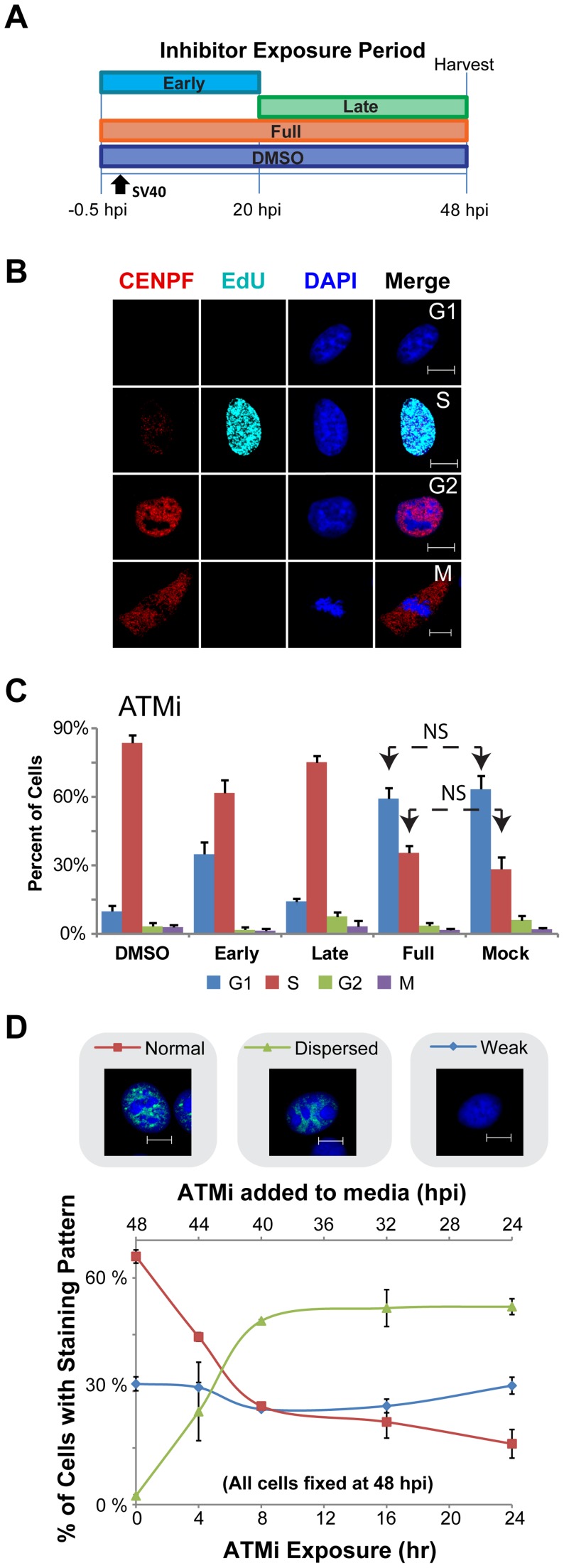
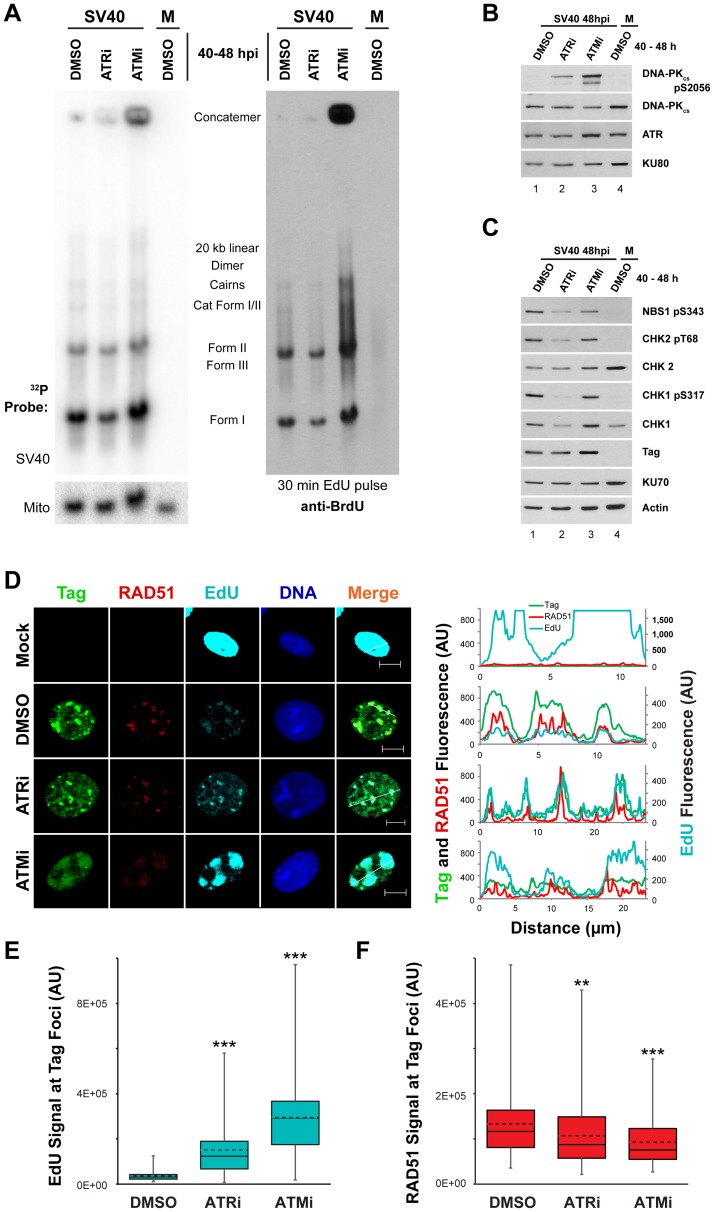
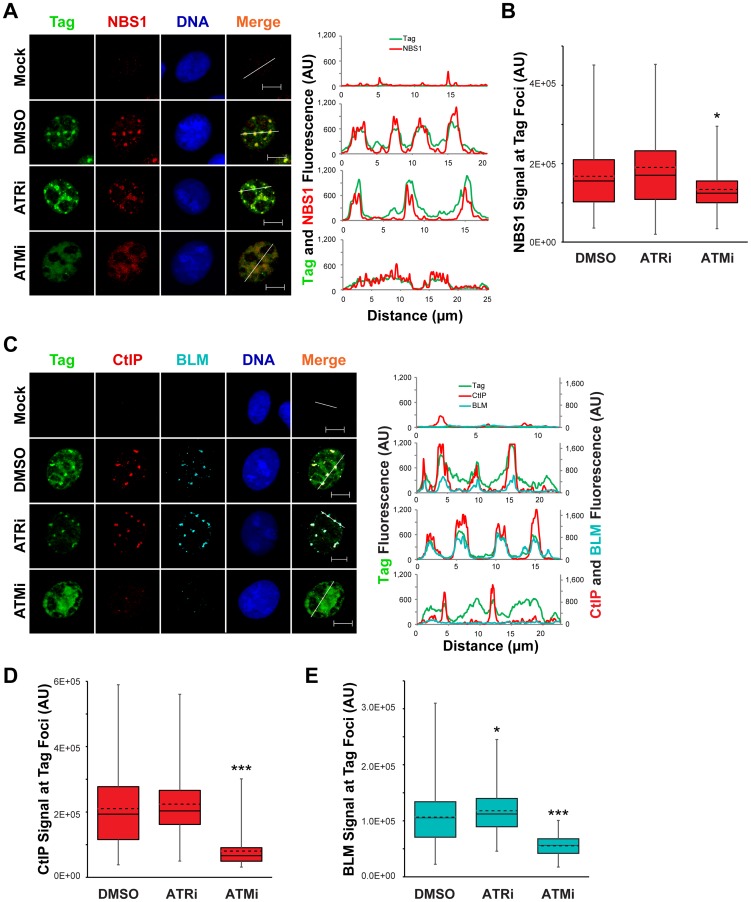
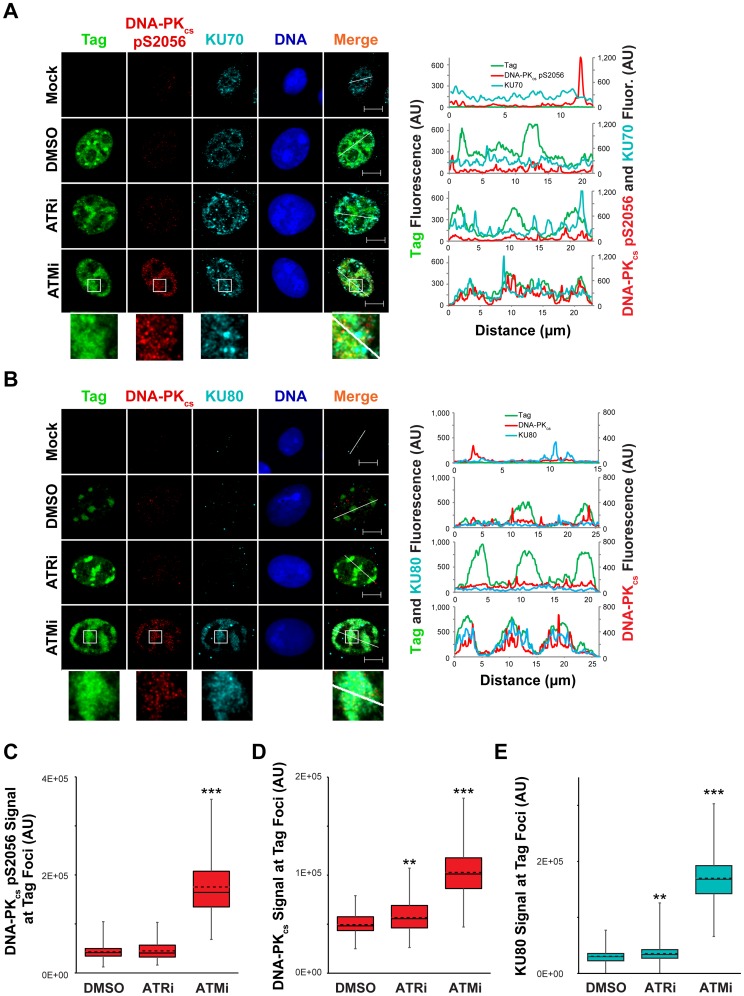
Simian virus 40 (SV40) and cellular DNA replication rely on host ATM and ATR DNA damage signaling kinases to facilitate DNA repair and elicit cell cycle arrest following DNA damage. During SV40 DNA replication, ATM kinase activity prevents concatemerization of the viral genome whereas ATR activity prevents accumulation of aberrant genomes resulting from breakage of a moving replication fork as it converges with a stalled fork. However, the repair pathways that ATM and ATR orchestrate to prevent these aberrant SV40 DNA replication products are unclear. Using two-dimensional gel electrophoresis and Southern blotting, we show that ATR kinase activity, but not DNA-PK(cs) kinase activity, facilitates some aspects of double strand break (DSB) repair when ATM is inhibited during SV40 infection. To clarify which repair factors associate with viral DNA replication centers, we examined the localization of DSB repair proteins in response to SV40 infection. Under normal conditions, viral replication centers exclusively associate with homology-directed repair (HDR) and do not colocalize with non-homologous end joining (NHEJ) factors. Following ATM inhibition, but not ATR inhibition, activated DNA-PK(cs) and KU70/80 accumulate at the viral replication centers while CtIP and BLM, proteins that initiate 5' to 3' end resection during HDR, become undetectable. Similar to what has been observed during cellular DSB repair in S phase, these data suggest that ATM kinase influences DSB repair pathway choice by preventing the recruitment of NHEJ factors to replicating viral DNA. These data may explain how ATM prevents concatemerization of the viral genome and promotes viral propagation. We suggest that inhibitors of DNA damage signaling and DNA repair could be used during infection to disrupt productive viral DNA replication.
猴病毒40(SV40)和细胞DNA复制依赖宿主的ATM和ATR DNA损伤信号激酶来促进DNA修复,并在DNA损伤后引发细胞周期停滞。在SV40 DNA复制过程中,ATM激酶活性可防止病毒基因组串联,而ATR活性可防止因移动的复制叉与停滞的叉汇合时断裂而产生的异常基因组积累。然而,ATM和ATR精心策划的防止这些异常SV40 DNA复制产物的修复途径尚不清楚。使用二维凝胶电泳和Southern印迹法,我们发现当在SV40感染期间抑制ATM时,ATR激酶活性而非DNA-PK(cs)激酶活性促进了双链断裂(DSB)修复的某些方面。为了阐明哪些修复因子与病毒DNA复制中心相关,我们研究了响应SV40感染时DSB修复蛋白的定位。在正常情况下,病毒复制中心仅与同源定向修复(HDR)相关,且不与非同源末端连接(NHEJ)因子共定位。在抑制ATM而非ATR后,活化的DNA-PK(cs)和KU70/80在病毒复制中心积累,而在HDR期间启动5'至3'末端切除的蛋白CtIP和BLM则无法检测到。与在细胞S期DSB修复过程中观察到的情况类似,这些数据表明ATM激酶通过阻止NHEJ因子募集到复制的病毒DNA上来影响DSB修复途径的选择。这些数据可能解释了ATM如何防止病毒基因组串联并促进病毒传播。我们建议在感染期间可使用DNA损伤信号和DNA修复抑制剂来破坏有生产性的病毒DNA复制。