Helmholtz-Zentrum München - German Research Centre for Environmental Health (GmbH), Institute of Structural Biology , Munich 85764, Germany.
J Chem Inf Model. 2014 Dec 22;54(12):3320-9. doi: 10.1021/ci5005288. Epub 2014 Dec 9.
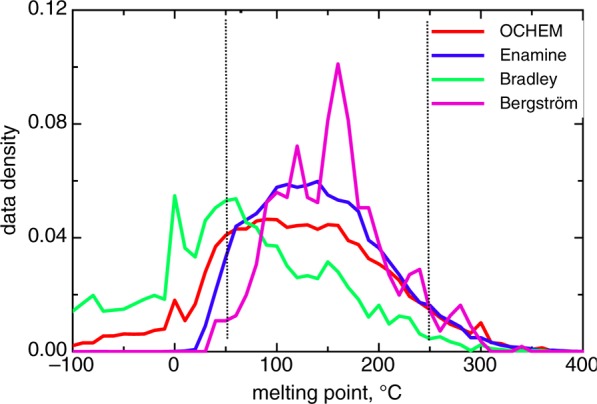
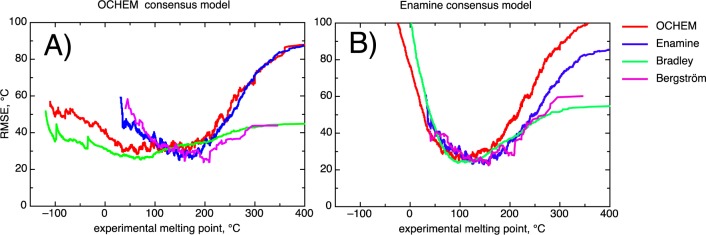
This article contributes a highly accurate model for predicting the melting points (MPs) of medicinal chemistry compounds. The model was developed using the largest published data set, comprising more than 47k compounds. The distributions of MPs in drug-like and drug lead sets showed that >90% of molecules melt within [50,250]°C. The final model calculated an RMSE of less than 33 °C for molecules from this temperature interval, which is the most important for medicinal chemistry users. This performance was achieved using a consensus model that performed calculations to a significantly higher accuracy than the individual models. We found that compounds with reactive and unstable groups were overrepresented among outlying compounds. These compounds could decompose during storage or measurement, thus introducing experimental errors. While filtering the data by removing outliers generally increased the accuracy of individual models, it did not significantly affect the results of the consensus models. Three analyzed distance to models did not allow us to flag molecules, which had MP values fell outside the applicability domain of the model. We believe that this negative result and the public availability of data from this article will encourage future studies to develop better approaches to define the applicability domain of models. The final model, MP data, and identified reactive groups are available online at http://ochem.eu/article/55638.
这篇文章贡献了一个高度准确的模型,用于预测药物化学化合物的熔点(MP)。该模型是使用最大的已发表数据集开发的,该数据集包含超过 47,000 种化合物。药物样和药物先导化合物集的 MPs 分布表明,>90%的分子在[50,250]°C 内熔化。最终模型计算出的温度间隔内分子的 RMSE 小于 33°C,这对药物化学用户来说是最重要的。这种性能是使用共识模型实现的,该模型的计算精度明显高于单个模型。我们发现,具有反应性和不稳定性基团的化合物在离群化合物中过度代表。这些化合物在储存或测量过程中可能会分解,从而引入实验误差。虽然通过去除离群值过滤数据通常会提高单个模型的准确性,但它不会显著影响共识模型的结果。我们分析的三个距离模型并没有使我们能够标记那些 MP 值落在模型应用域之外的分子。我们认为,这一负面结果以及本文中数据的公开可用性将鼓励未来的研究开发出更好的方法来定义模型的应用域。最终模型、MP 数据和识别出的反应性基团可在 http://ochem.eu/article/55638 上在线获取。