Dénes Judit, Swords Francesca, Rattenberry Eleanor, Stals Karen, Owens Martina, Cranston Treena, Xekouki Paraskevi, Moran Linda, Kumar Ajith, Wassif Christopher, Fersht Naomi, Baldeweg Stephanie E, Morris Damian, Lightman Stafford, Agha Amar, Rees Aled, Grieve Joan, Powell Michael, Boguszewski Cesar Luiz, Dutta Pinaki, Thakker Rajesh V, Srirangalingam Umasuthan, Thompson Chris J, Druce Maralyn, Higham Claire, Davis Julian, Eeles Rosalind, Stevenson Mark, O'Sullivan Brendan, Taniere Phillipe, Skordilis Kassiani, Gabrovska Plamena, Barlier Anne, Webb Susan M, Aulinas Anna, Drake William M, Bevan John S, Preda Cristina, Dalantaeva Nadezhda, Ribeiro-Oliveira Antônio, Garcia Isabel Tena, Yordanova Galina, Iotova Violeta, Evanson Jane, Grossman Ashley B, Trouillas Jacqueline, Ellard Sian, Stratakis Constantine A, Maher Eamonn R, Roncaroli Federico, Korbonits Márta
Department of Endocrinology (J.D., U.S., M.D., P.G., W.M.D., M.K.), Barts and the London School of Medicine, Queen Mary University of London, London EC1M 6BQ, United Kingdom; Semmelweis University, School of PhD studies, Doctoral School of Clinical Medicine, Budapest, Hungary (J.D.), Endocrinology Directorate (F.S.), Norfolk and Norwich University Hospital, Norwich NR4 7UZ, United Kingdom; Department of Medical and Molecular Genetics (E.R., E.R.M.), University of Birmingham, Birmingham B15 2TT, United Kingdom; Department of Molecular Genetics (K.S., M.O., S.E.), Royal Devon and Exeter National Health Service Foundation Trust, Exeter EX2 5DW, United Kingdom; University of Exeter Medical School (S.E.), Exeter EX4 4PY, United Kingdom; Oxford Medical Genetics Laboratories (T.C.), Oxford University Hospitals National Health Service Trust, The Churchill Hospital, Oxford OX3 7LJ, United Kingdom; Section on Endocrinology and Genetics (P.X., C.A.S.) and Section on Molecular Dysmorphology (C.W.), Eunice Kennedy Shriver Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland 20892; Electron Microscopy Unit (L.M.), Department Histopathology, Charing Cross Hospital, Imperial College Healthcare National Health Service Trust, London W6 8RF, United Kingdom; Department of Clinical Genetics (A.K.), Great Ormond Street Hospital, London WC1N 1LE, United Kingdom; Departments of Oncology (N.F.) and Endocrinology (S.E.B.), University College London Hospitals, London WC1E 6BT, United Kingdom; Department of Diabetes and Endocrinology (D.M.), The Ipswich Hospital National Health Service Trust, Ipswich IP4 5PD, United Kingdom; Henry Wellcome Laboratories for Integrative Neuroscience and Endocrinology (S.L.), University of Bristol, Bristol BS1 3NY, United Kingdom; Department of Endocrinology (A.Ag., C.J.T.), Beaumont Hospital, Dublin 9, Ireland; Institute of Molecular and Experimental Medicine (A.R.), Cardiff University, Cardiff CF10 3US, United Kingd
J Clin Endocrinol Metab. 2015 Mar;100(3):E531-41. doi: 10.1210/jc.2014-3399. Epub 2014 Dec 12.
Pituitary adenomas and pheochromocytomas/paragangliomas (pheo/PGL) can occur in the same patient or in the same family. Coexistence of the two diseases could be due to either a common pathogenic mechanism or a coincidence.
The objective of the investigation was to study the possible coexistence of pituitary adenoma and pheo/PGL.
Thirty-nine cases of sporadic or familial pheo/PGL and pituitary adenomas were investigated. Known pheo/PGL genes (SDHA-D, SDHAF2, RET, VHL, TMEM127, MAX, FH) and pituitary adenoma genes (MEN1, AIP, CDKN1B) were sequenced using next generation or Sanger sequencing. Loss of heterozygosity study and pathological studies were performed on the available tumor samples.
The study was conducted at university hospitals.
Thirty-nine patients with sporadic of familial pituitary adenoma and pheo/PGL participated in the study.
Outcomes included genetic screening and clinical characteristics.
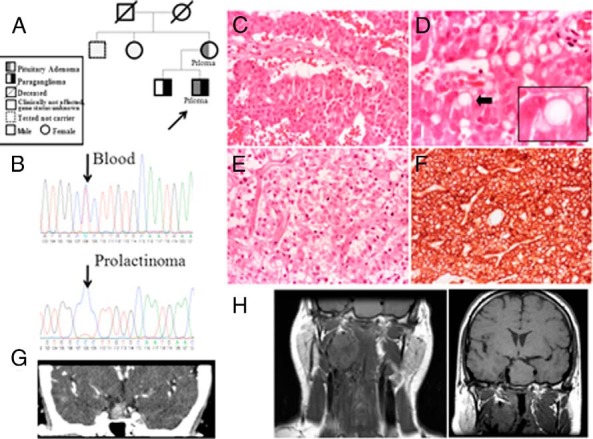
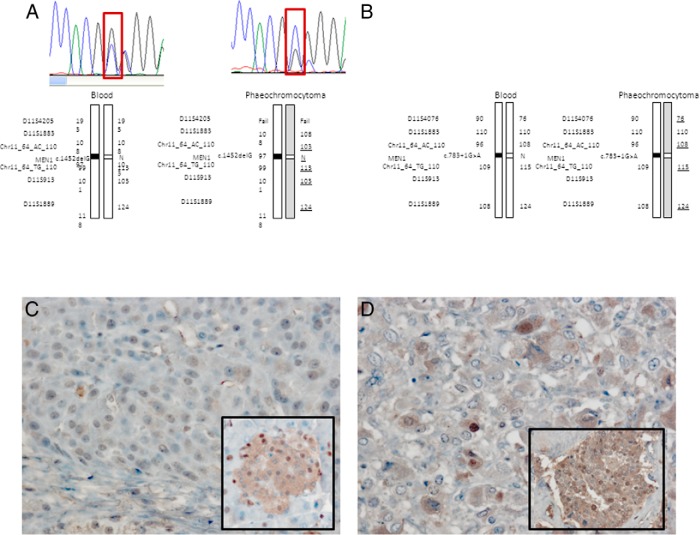
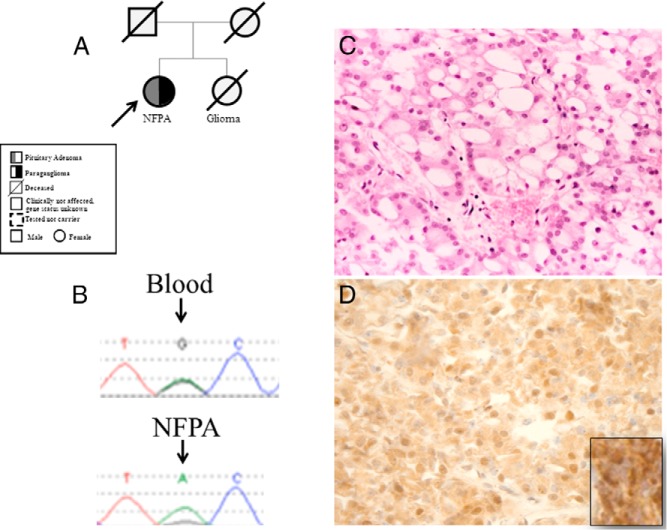
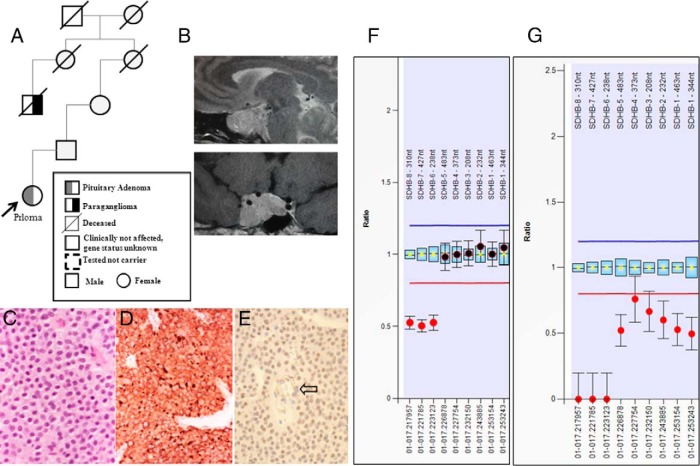
Eleven germline mutations (five SDHB, one SDHC, one SDHD, two VHL, and two MEN1) and four variants of unknown significance (two SDHA, one SDHB, and one SDHAF2) were identified in the studied genes in our patient cohort. Tumor tissue analysis identified LOH at the SDHB locus in three pituitary adenomas and loss of heterozygosity at the MEN1 locus in two pheochromocytomas. All the pituitary adenomas of patients affected by SDHX alterations have a unique histological feature not previously described in this context.
Mutations in the genes known to cause pheo/PGL can rarely be associated with pituitary adenomas, whereas mutation in a gene predisposing to pituitary adenomas (MEN1) can be associated with pheo/PGL. Our findings suggest that genetic testing should be considered in all patients or families with the constellation of pheo/PGL and a pituitary adenoma.
垂体腺瘤与嗜铬细胞瘤/副神经节瘤(嗜铬细胞瘤/副神经节瘤)可发生于同一患者或同一家族。这两种疾病的共存可能是由于共同的致病机制或巧合。
本研究旨在探讨垂体腺瘤与嗜铬细胞瘤/副神经节瘤可能共存的情况。
对39例散发性或家族性嗜铬细胞瘤/副神经节瘤及垂体腺瘤患者进行研究。使用二代测序或桑格测序对已知的嗜铬细胞瘤/副神经节瘤基因(SDHA - D、SDHAF2、RET、VHL、TMEM127、MAX、FH)和垂体腺瘤基因(MEN1、AIP、CDKN1B)进行测序。对可用的肿瘤样本进行杂合性缺失研究和病理研究。
本研究在大学医院进行。
39例散发性或家族性垂体腺瘤及嗜铬细胞瘤/副神经节瘤患者参与了本研究。
在我们的患者队列中,在所研究的基因中鉴定出11种胚系突变(5种SDHB、1种SDHC、1种SDHD、2种VHL和2种MEN1)以及4种意义不明的变异(2种SDHA、1种SDHB和1种SDHAF2)。肿瘤组织分析在3例垂体腺瘤中发现SDHB基因座的杂合性缺失,在2例嗜铬细胞瘤中发现MEN1基因座的杂合性缺失。受SDHX改变影响的患者的所有垂体腺瘤都有在此背景下以前未描述过的独特组织学特征。
已知导致嗜铬细胞瘤/副神经节瘤的基因突变很少与垂体腺瘤相关,而倾向于导致垂体腺瘤的基因(MEN1)突变可与嗜铬细胞瘤/副神经节瘤相关。我们的研究结果表明,对于所有患有嗜铬细胞瘤/副神经节瘤和垂体腺瘤的患者或家族,都应考虑进行基因检测。