Mu John C, Mohiyuddin Marghoob, Li Jian, Bani Asadi Narges, Gerstein Mark B, Abyzov Alexej, Wong Wing H, Lam Hugo Y K
Department of Electrical Engineering, Stanford University, Stanford, CA 94035, USA, Department of Bioinformatics, Bina Technologies, Redwood City, CA 94065, USA, Program in Computational Biology and Bioinformatics, Yale University, New Haven, CT 06520, USA, Mayo Clinics, Department of Health Sciences Research, Rochester, MN 55902, USA, Department of Statistics, Stanford University, Stanford, CA 94035, USA and Department of Health Research and Policy, Stanford University, Stanford, CA 94035, USA Department of Electrical Engineering, Stanford University, Stanford, CA 94035, USA, Department of Bioinformatics, Bina Technologies, Redwood City, CA 94065, USA, Program in Computational Biology and Bioinformatics, Yale University, New Haven, CT 06520, USA, Mayo Clinics, Department of Health Sciences Research, Rochester, MN 55902, USA, Department of Statistics, Stanford University, Stanford, CA 94035, USA and Department of Health Research and Policy, Stanford University, Stanford, CA 94035, USA.
Department of Electrical Engineering, Stanford University, Stanford, CA 94035, USA, Department of Bioinformatics, Bina Technologies, Redwood City, CA 94065, USA, Program in Computational Biology and Bioinformatics, Yale University, New Haven, CT 06520, USA, Mayo Clinics, Department of Health Sciences Research, Rochester, MN 55902, USA, Department of Statistics, Stanford University, Stanford, CA 94035, USA and Department of Health Research and Policy, Stanford University, Stanford, CA 94035, USA.
Bioinformatics. 2015 May 1;31(9):1469-71. doi: 10.1093/bioinformatics/btu828. Epub 2014 Dec 17.
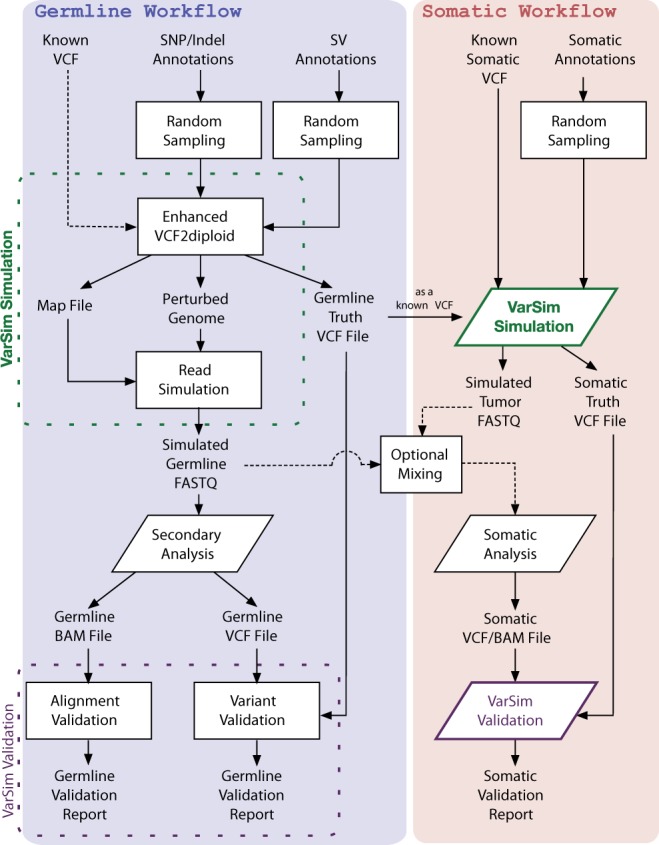
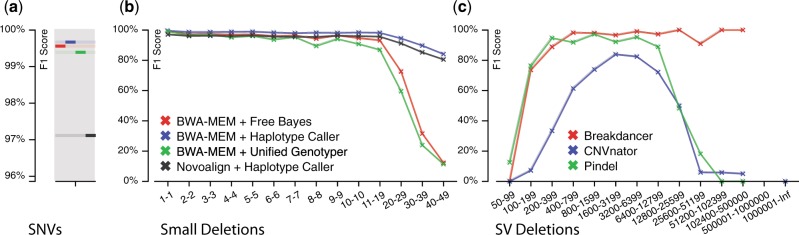
VarSim is a framework for assessing alignment and variant calling accuracy in high-throughput genome sequencing through simulation or real data. In contrast to simulating a random mutation spectrum, it synthesizes diploid genomes with germline and somatic mutations based on a realistic model. This model leverages information such as previously reported mutations to make the synthetic genomes biologically relevant. VarSim simulates and validates a wide range of variants, including single nucleotide variants, small indels and large structural variants. It is an automated, comprehensive compute framework supporting parallel computation and multiple read simulators. Furthermore, we developed a novel map data structure to validate read alignments, a strategy to compare variants binned in size ranges and a lightweight, interactive, graphical report to visualize validation results with detailed statistics. Thus far, it is the most comprehensive validation tool for secondary analysis in next generation sequencing.
Code in Java and Python along with instructions to download the reads and variants is at http://bioinform.github.io/varsim.
Supplementary data are available at Bioinformatics online.
VarSim是一个通过模拟或真实数据来评估高通量基因组测序中比对和变异检测准确性的框架。与模拟随机突变谱不同,它基于一个现实模型合成具有种系和体细胞突变的二倍体基因组。该模型利用诸如先前报道的突变等信息,使合成基因组具有生物学相关性。VarSim模拟并验证广泛的变异,包括单核苷酸变异、小插入缺失和大结构变异。它是一个支持并行计算和多个读取模拟器的自动化、综合性计算框架。此外,我们开发了一种新颖的映射数据结构来验证读取比对,一种按大小范围对变异进行分组比较的策略,以及一个轻量级、交互式的图形报告,以可视化带有详细统计信息的验证结果。到目前为止,它是下一代测序中二级分析最全面的验证工具。
Java和Python代码以及下载读取和变异的说明可在http://bioinform.github.io/varsim获取。
补充数据可在《生物信息学》在线获取。