Chen Lin-xing, Hu Min, Huang Li-nan, Hua Zheng-shuang, Kuang Jia-liang, Li Sheng-jin, Shu Wen-sheng
State Key Laboratory of Biocontrol, Key Laboratory of Biodiversity Dynamics and Conservation of Guangdong Higher Education Institutes, College of Ecology and Evolution, Sun Yat-sen University, Guangzhou, People's Republic of China.
ISME J. 2015 Jul;9(7):1579-92. doi: 10.1038/ismej.2014.245. Epub 2014 Dec 23.
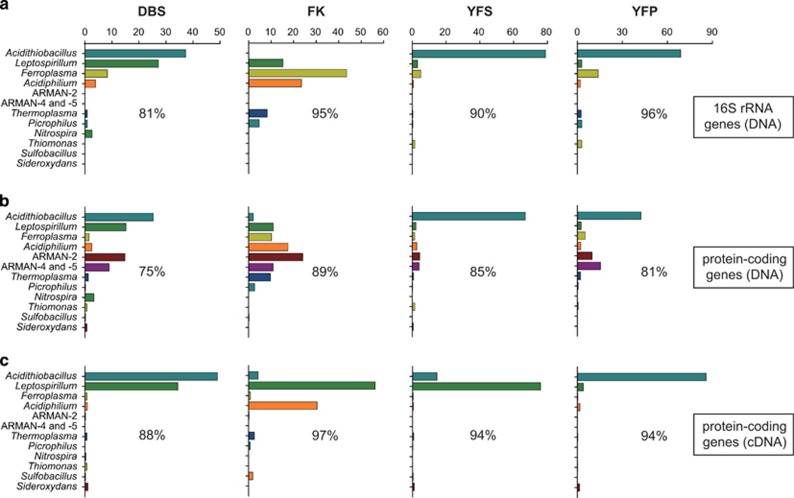
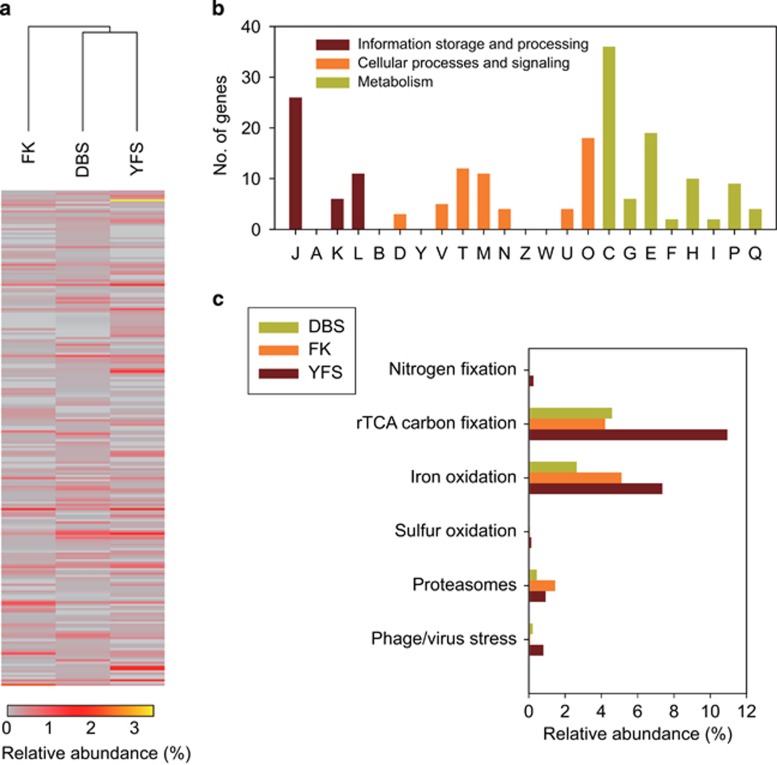
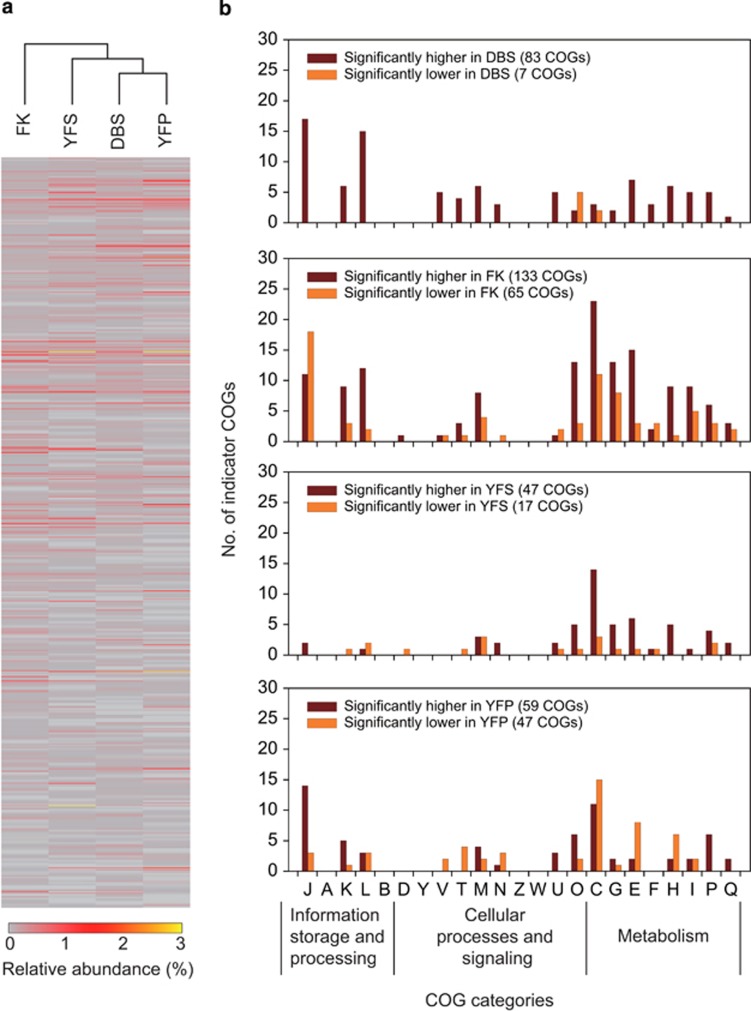
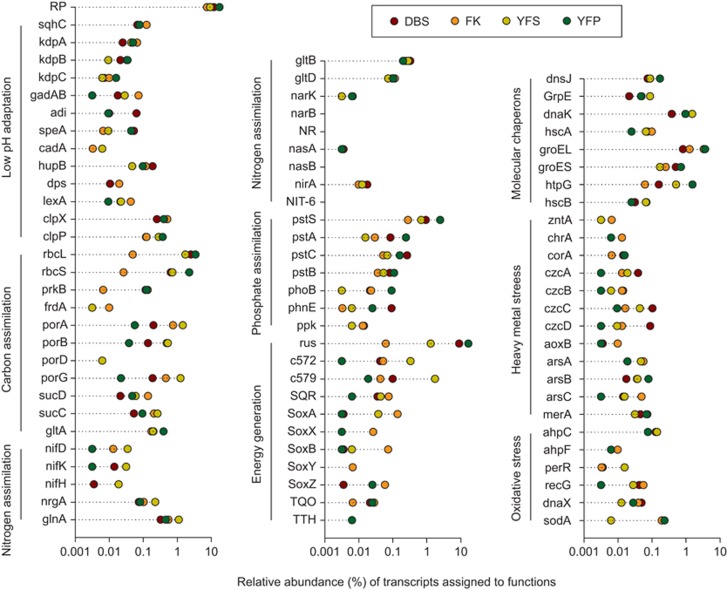
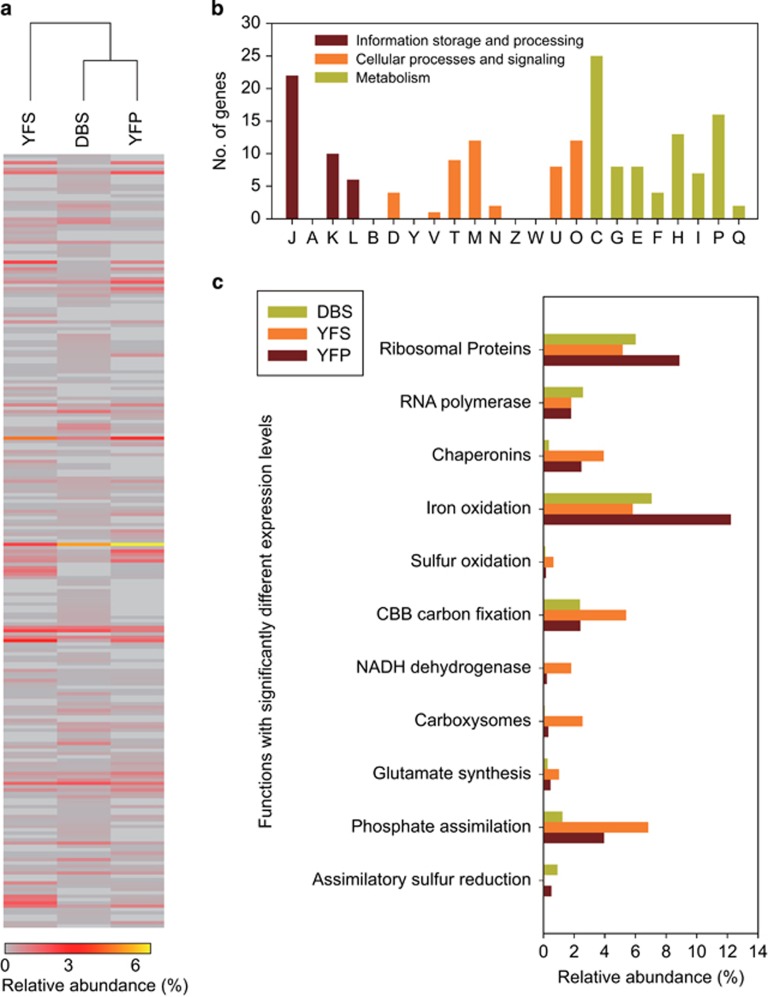
The microbial communities in acid mine drainage have been extensively studied to reveal their roles in acid generation and adaption to this environment. Lacking, however, are integrated community- and organism-wide comparative gene transcriptional analyses that could reveal the response and adaptation mechanisms of these extraordinary microorganisms to different environmental conditions. In this study, comparative metagenomics and metatranscriptomics were performed on microbial assemblages collected from four geochemically distinct acid mine drainage (AMD) sites. Taxonomic analysis uncovered unexpectedly high microbial biodiversity of these extremely acidophilic communities, and the abundant taxa of Acidithiobacillus, Leptospirillum and Acidiphilium exhibited high transcriptional activities. Community-wide comparative analyses clearly showed that the AMD microorganisms adapted to the different environmental conditions via regulating the expression of genes involved in multiple in situ functional activities, including low-pH adaptation, carbon, nitrogen and phosphate assimilation, energy generation, environmental stress resistance, and other functions. Organism-wide comparative analyses of the active taxa revealed environment-dependent gene transcriptional profiles, especially the distinct strategies used by Acidithiobacillus ferrivorans and Leptospirillum ferrodiazotrophum in nutrients assimilation and energy generation for survival under different conditions. Overall, these findings demonstrate that the gene transcriptional profiles of AMD microorganisms are closely related to the site physiochemical characteristics, providing clues into the microbial response and adaptation mechanisms in the oligotrophic, extremely acidic environments.
为揭示酸性矿山排水中的微生物群落产生酸性及适应该环境的作用,人们已对其展开广泛研究。然而,尚缺乏整合群落和全生物体水平的比较基因转录分析,而这种分析能够揭示这些特殊微生物对不同环境条件的响应及适应机制。在本研究中,我们对从四个地球化学特征不同的酸性矿山排水(AMD)位点采集的微生物群落进行了比较宏基因组学和宏转录组学研究。分类学分析意外发现这些极端嗜酸群落具有极高的微生物多样性,嗜酸氧化硫硫杆菌、钩端螺旋菌和嗜酸菌等优势类群表现出较高的转录活性。全群落比较分析清楚地表明,AMD微生物通过调控参与多种原位功能活动的基因表达来适应不同环境条件,这些活动包括低pH适应、碳、氮和磷同化、能量产生、环境应激抗性及其他功能。对活跃类群的全生物体比较分析揭示了依赖环境的基因转录谱,特别是嗜酸氧化亚铁硫杆菌和嗜铁脱氮钩端螺旋菌在不同条件下生存时用于养分同化和能量产生的独特策略。总体而言,这些发现表明AMD微生物的基因转录谱与位点理化特征密切相关,为贫营养、极端酸性环境中的微生物响应和适应机制提供了线索。