Heydarian Mohammad, Luperchio Teresa Romeo, Cutler Jevon, Mitchell Christopher J, Kim Min-Sik, Pandey Akhilesh, Sollner-Webb Barbara, Reddy Karen
Johns Hopkins University, Department of Biological Chemistry, 725 North Wolfe Street, Baltimore, USA ; Johns Hopkins University, Center for Epigenetics, 855 North Wolfe Street, Baltimore, USA.
Johns Hopkins University, Department of Biological Chemistry, 725 North Wolfe Street, Baltimore, USA ; Johns Hopkins University, Center for Epigenetics, 855 North Wolfe Street, Baltimore, USA ; Johns Hopkins University, McKusick-Nathans Institute of Genetic Medicine, 733 North, Broadway Avenue, Baltimore, USA.
J Proteomics Bioinform. 2014 Feb 17;7. doi: 10.4172/jpb.1000302.
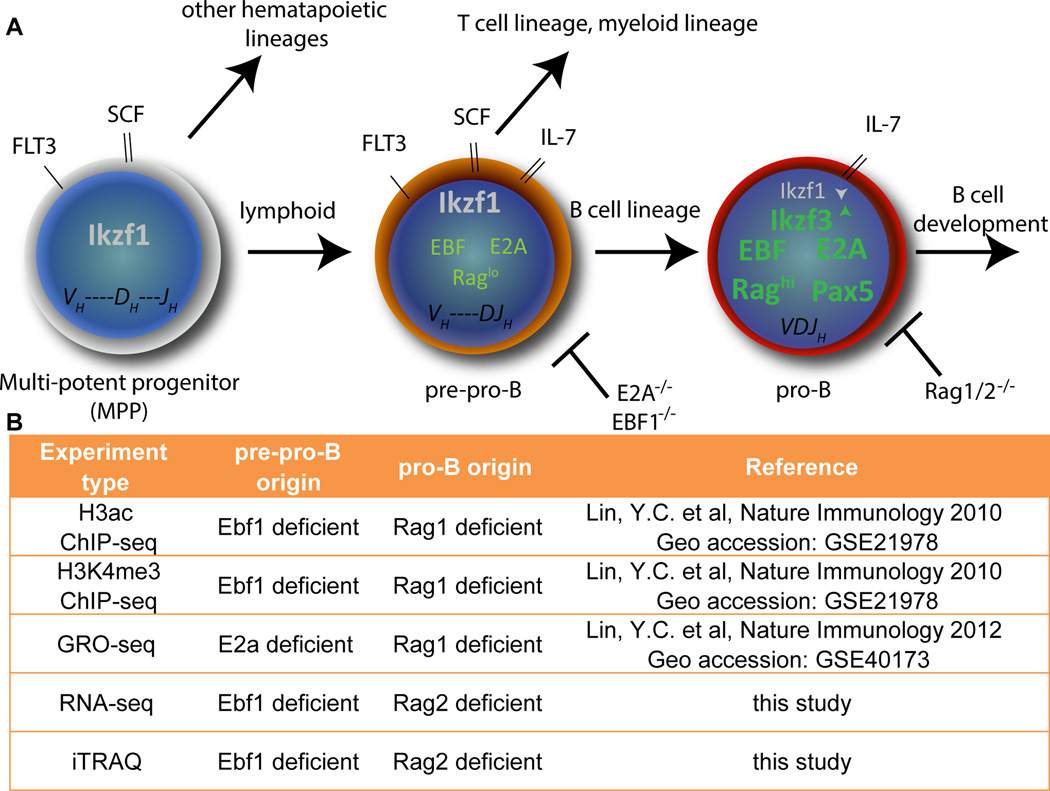
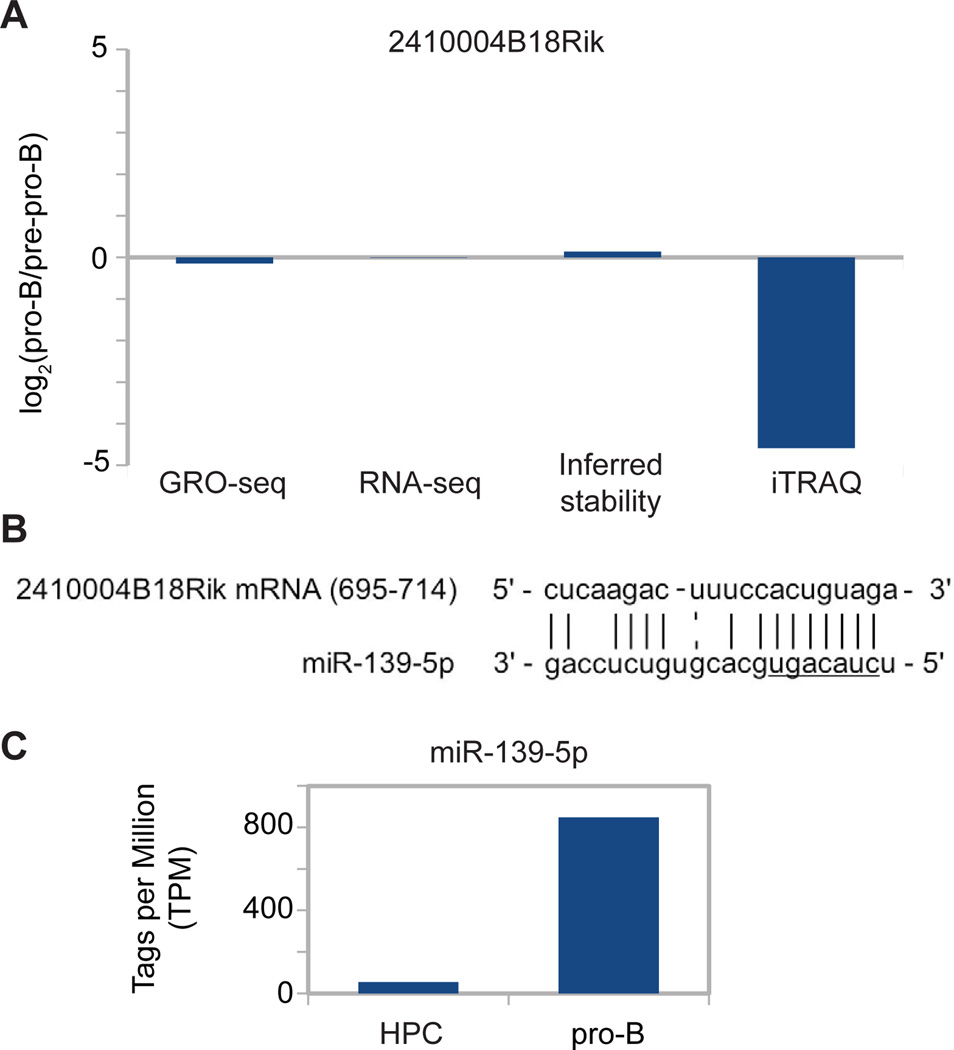
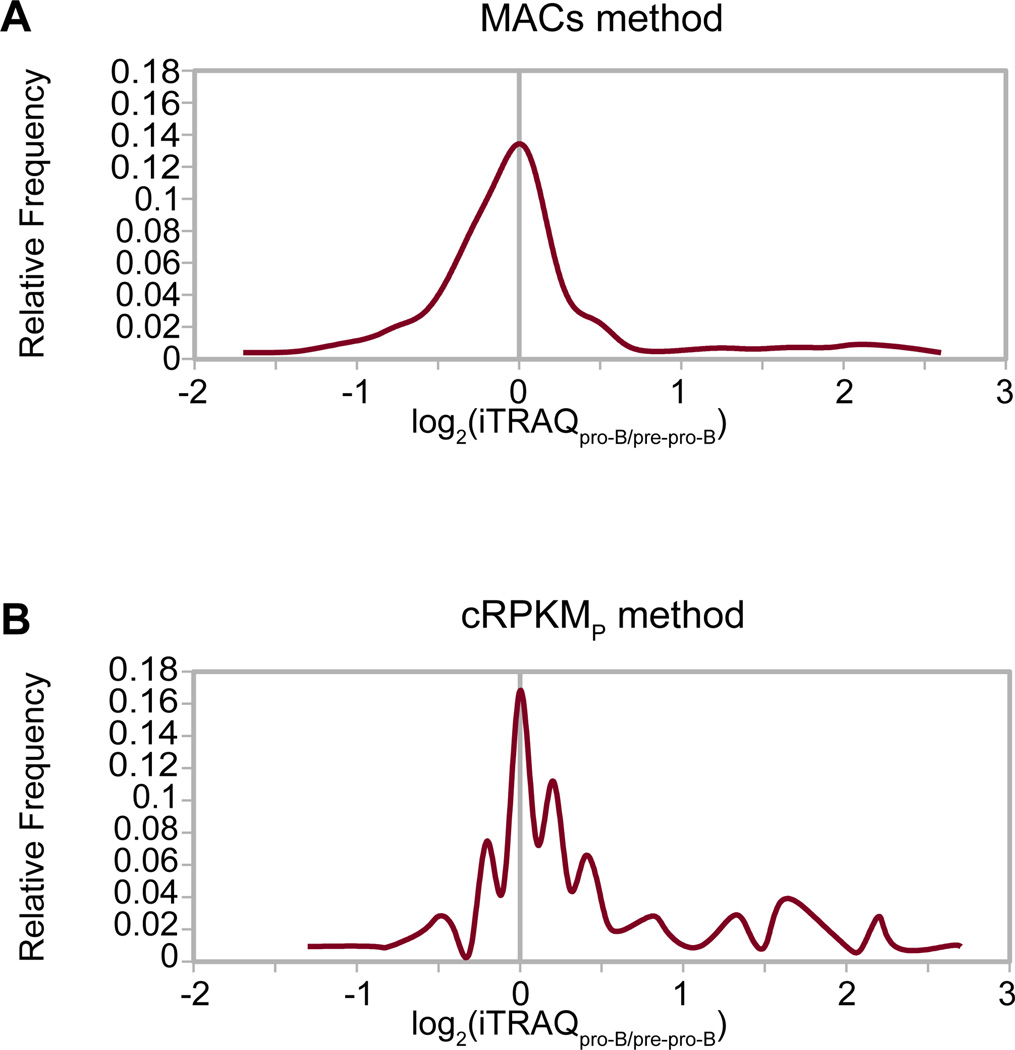
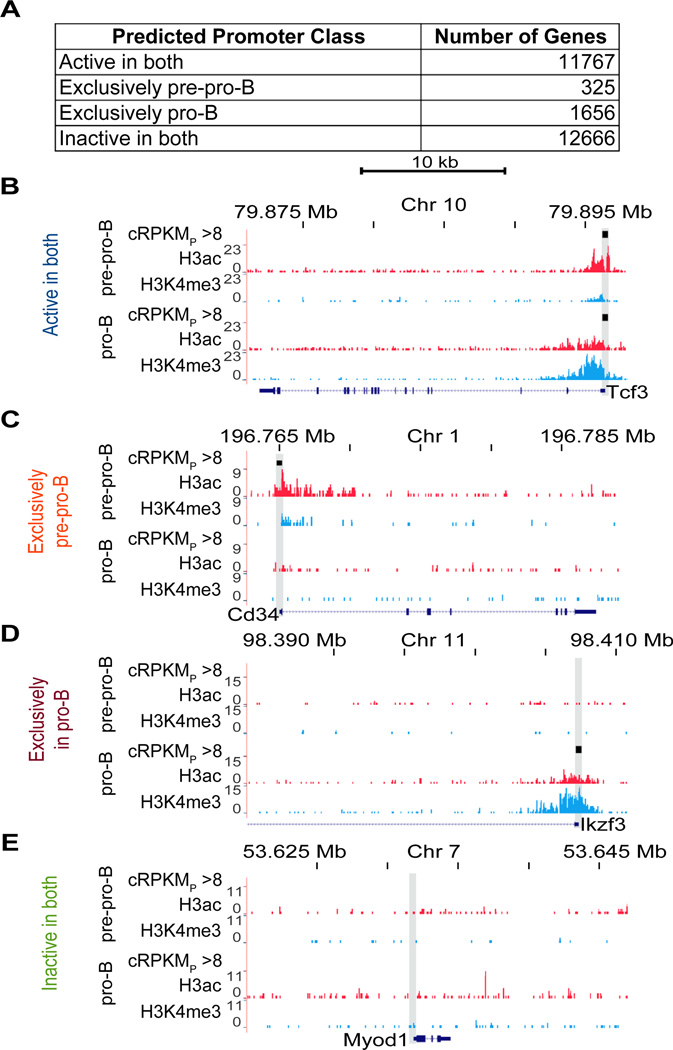
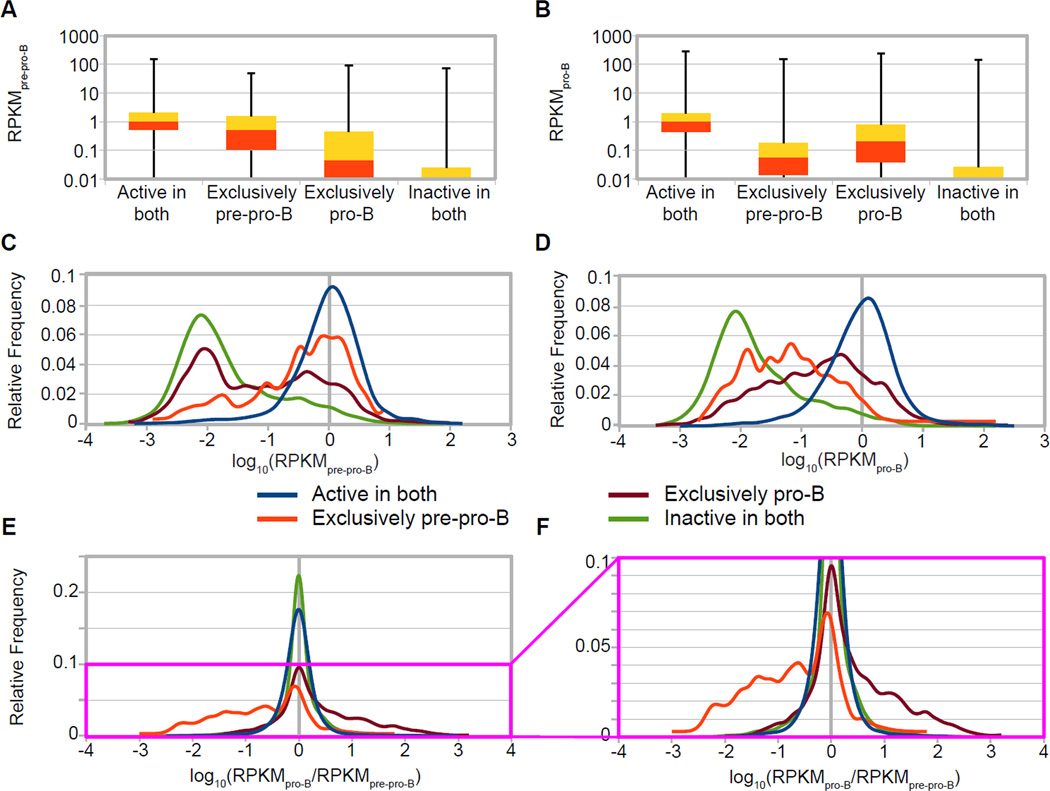
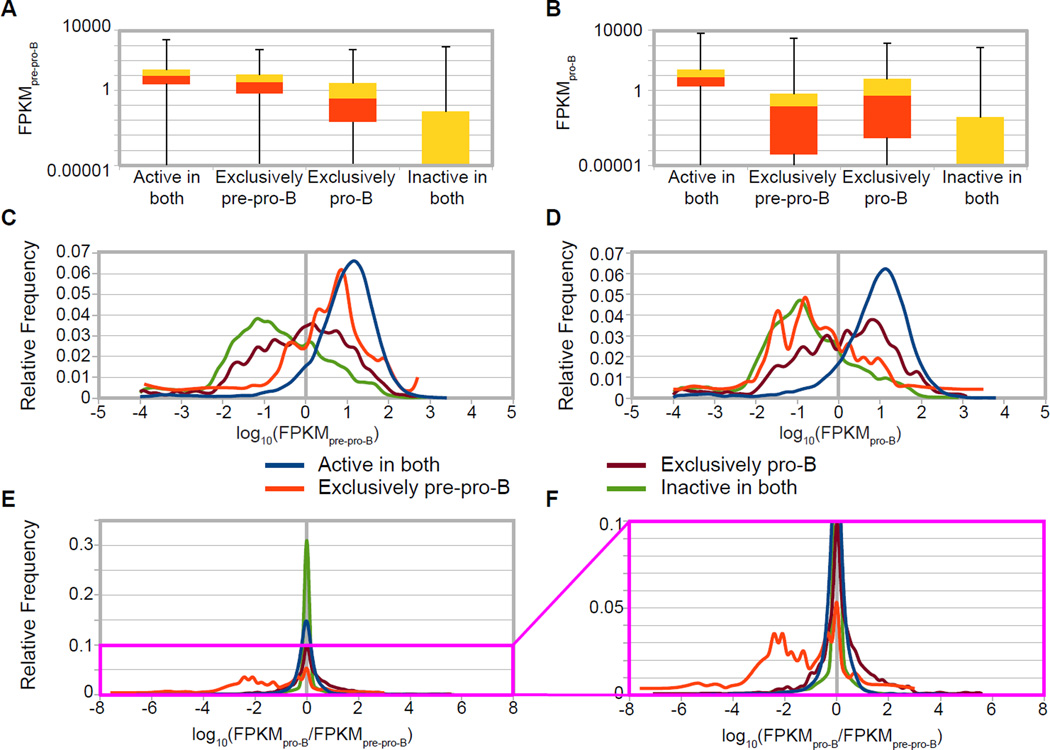
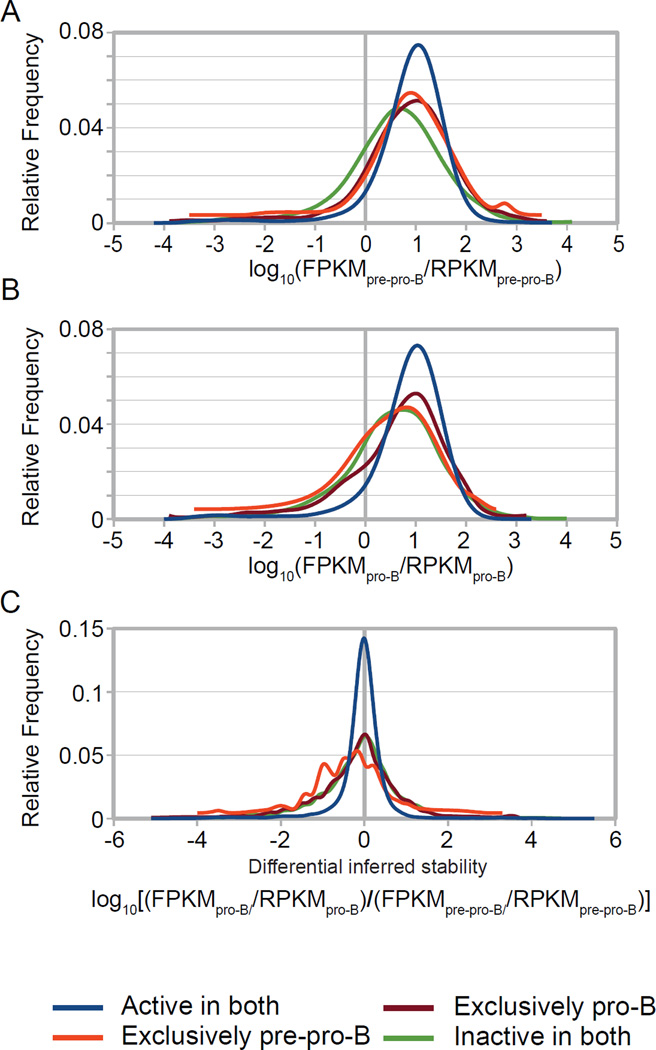
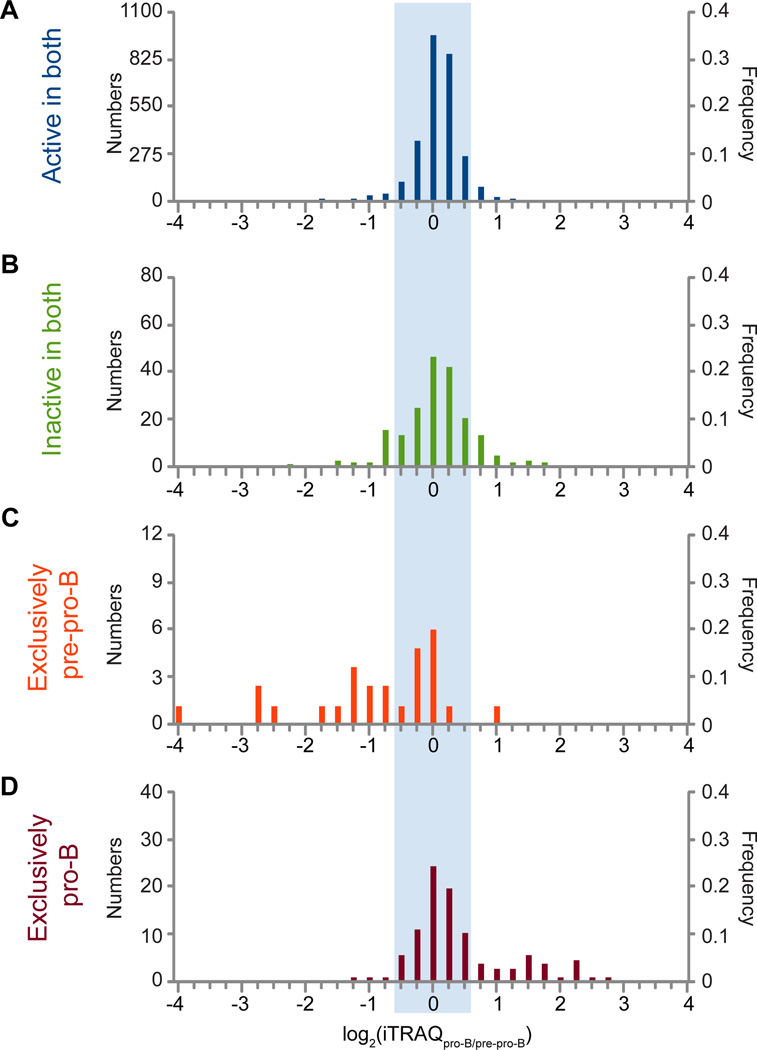
An increasingly common method for predicting gene activity is genome-wide chromatin immuno-precipitation of 'active' chromatin modifications followed by massively parallel sequencing (ChIP-seq). In order to understand better the relationship between developmentally regulated chromatin landscapes and regulation of early B cell development, we determined how differentially active promoter regions were able to predict relative RNA and protein levels at the pre-pro-B and pro-B stages. Herein, we describe a novel ChIP-seq quantification method (cRPKM) to identify active promoters and a multi-omics approach that compares promoter chromatin status with ongoing active transcription (GRO-seq), steady state mRNA (RNA-seq), inferred mRNA stability, and relative proteome abundance measurements (iTRAQ). We demonstrate that active chromatin modifications at promoters are good indicators of transcription and steady state mRNA levels. Moreover, we found that promoters with active chromatin modifications exclusively in one of these cell states frequently predicted the differential abundance of proteins. However, we found that many genes whose promoters have non-differential but active chromatin modifications also displayed changes in abundance of their cognate proteins. As expected, this large class of developmentally and differentially regulated proteins that was uncoupled from chromatin status used mostly post-transcriptional mechanisms. Strikingly, the most differentially abundant protein in our B-cell development system, 2410004B18Rik, was regulated by a post-transcriptional mechanism, which further analyses indicated was mediated by a micro-RNA. These data highlight how this integrated multi-omics data set can be a useful resource in uncovering regulatory mechanisms. This data can be accessed at: https://usegalaxy.org/u/thereddylab/p/prediction-of-gene-activity-based-on-an-integrative-multi-omics-analysis.
一种越来越常见的预测基因活性的方法是对“活性”染色质修饰进行全基因组染色质免疫沉淀,然后进行大规模平行测序(ChIP-seq)。为了更好地理解发育调控的染色质景观与早期B细胞发育调控之间的关系,我们确定了差异活跃的启动子区域如何能够预测前B祖细胞和B前体细胞阶段的相对RNA和蛋白质水平。在此,我们描述了一种用于识别活性启动子的新型ChIP-seq定量方法(cRPKM)以及一种多组学方法,该方法将启动子染色质状态与正在进行的活性转录(GRO-seq)、稳态mRNA(RNA-seq)、推断的mRNA稳定性和相对蛋白质组丰度测量(iTRAQ)进行比较。我们证明启动子处的活性染色质修饰是转录和稳态mRNA水平的良好指标。此外我们发现,仅在其中一种细胞状态下具有活性染色质修饰的启动子经常预测蛋白质的差异丰度。然而我们发现,许多启动子具有非差异但活性染色质修饰的基因,其同源蛋白质的丰度也发生了变化。正如预期的那样,这类大量由发育调控且差异调控的蛋白质与染色质状态解偶联,主要使用转录后机制。引人注目的是,我们的B细胞发育系统中差异丰度最高的蛋白质2410004B18Rik是由转录后机制调控的,进一步分析表明这是由一种微小RNA介导的。这些数据突出了这个整合的多组学数据集如何能够成为揭示调控机制的有用资源。该数据可从以下网址获取:https://usegalaxy.org/u/thereddylab/p/prediction-of-gene-activity-based-on-an-integrative-multi-omics-analysis 。