Hung Che-Lun, Chen Wen-Pei, Hua Guan-Jie, Zheng Huiru, Tsai Suh-Jen Jane, Lin Yaw-Ling
Department of Computer Science and Communication Engineering, Providence University, Taichung 43301, Taiwan.
Department of Applied Chemistry, Providence University, Taiwan 43301, Taiwan.
Int J Mol Sci. 2015 Jan 5;16(1):1096-110. doi: 10.3390/ijms16011096.
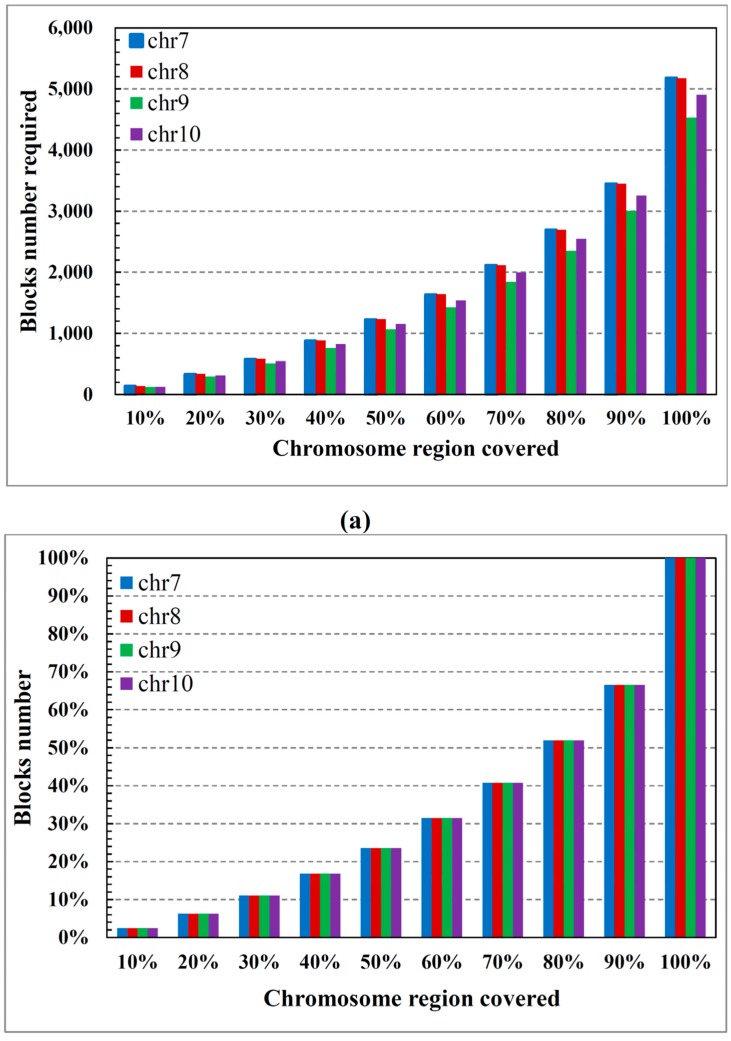
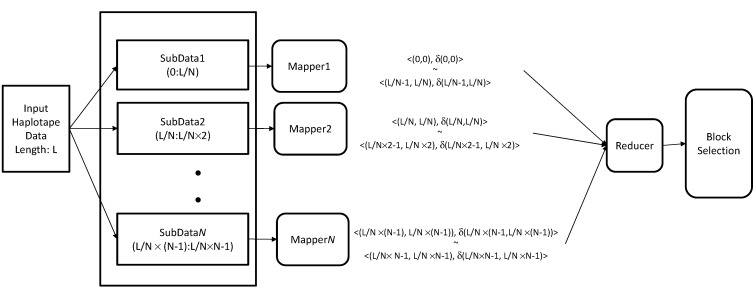
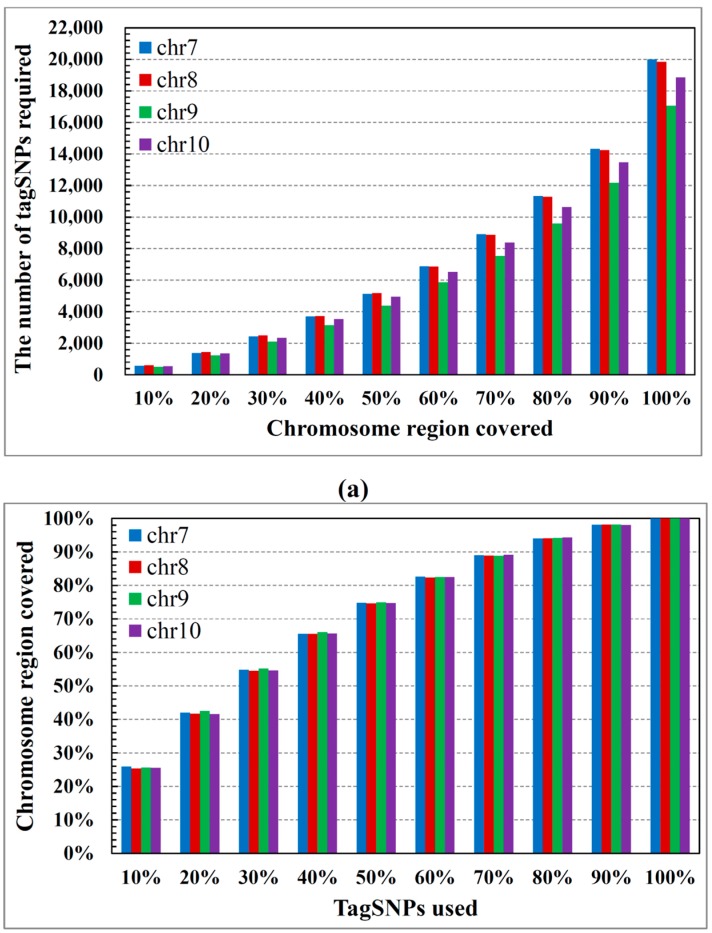
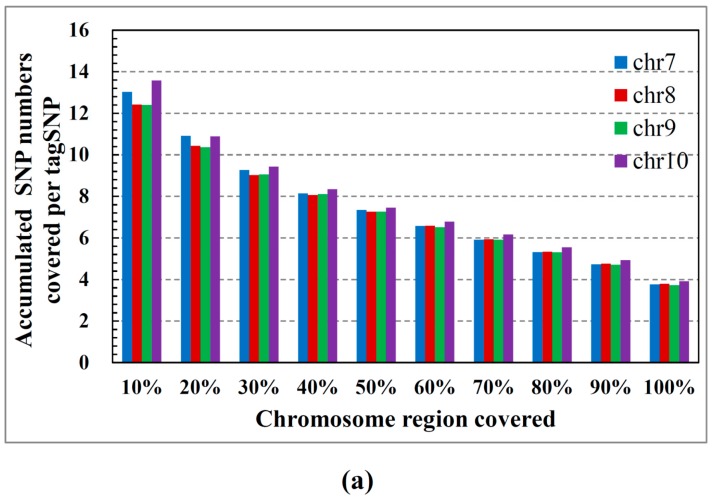
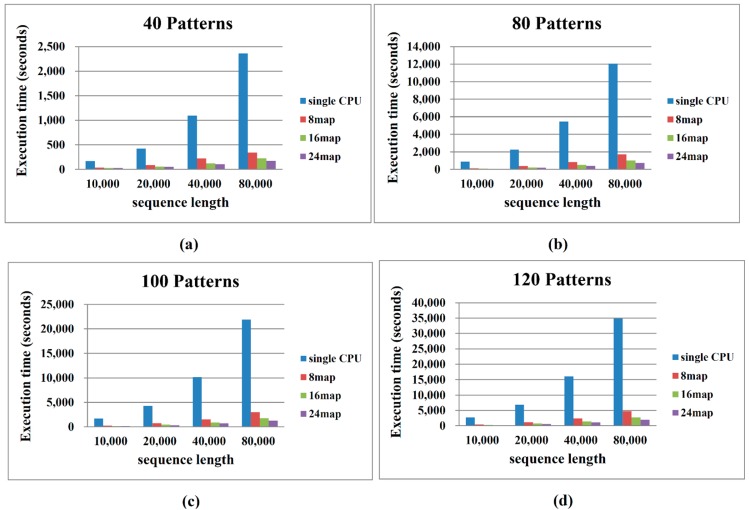
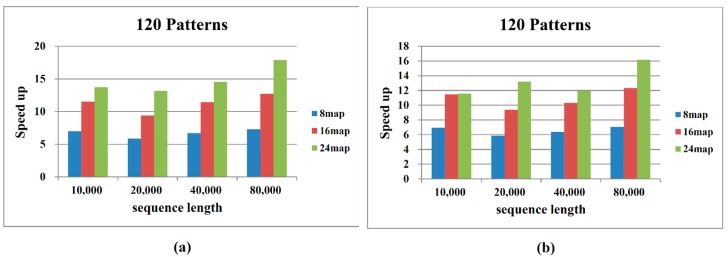
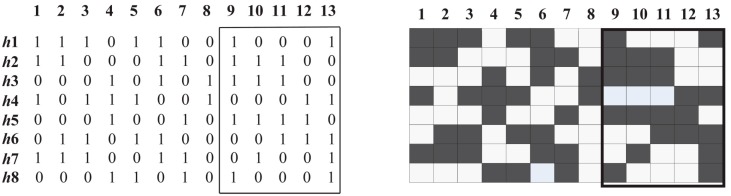
Single nucleotide polymorphisms (SNPs) play a fundamental role in human genetic variation and are used in medical diagnostics, phylogeny construction, and drug design. They provide the highest-resolution genetic fingerprint for identifying disease associations and human features. Haplotypes are regions of linked genetic variants that are closely spaced on the genome and tend to be inherited together. Genetics research has revealed SNPs within certain haplotype blocks that introduce few distinct common haplotypes into most of the population. Haplotype block structures are used in association-based methods to map disease genes. In this paper, we propose an efficient algorithm for identifying haplotype blocks in the genome. In chromosomal haplotype data retrieved from the HapMap project website, the proposed algorithm identified longer haplotype blocks than an existing algorithm. To enhance its performance, we extended the proposed algorithm into a parallel algorithm that copies data in parallel via the Hadoop MapReduce framework. The proposed MapReduce-paralleled combinatorial algorithm performed well on real-world data obtained from the HapMap dataset; the improvement in computational efficiency was proportional to the number of processors used.
单核苷酸多态性(SNPs)在人类遗传变异中起着基础性作用,并被用于医学诊断、系统发育构建和药物设计。它们为识别疾病关联和人类特征提供了最高分辨率的基因指纹。单倍型是基因组上紧密相连的连锁遗传变异区域,倾向于一起遗传。遗传学研究揭示了某些单倍型块内的单核苷酸多态性,这些单核苷酸多态性在大多数人群中引入了少数几种不同的常见单倍型。单倍型块结构被用于基于关联的方法来定位疾病基因。在本文中,我们提出了一种在基因组中识别单倍型块的高效算法。在从HapMap项目网站检索到的染色体单倍型数据中,该算法识别出的单倍型块比现有算法识别出的更长。为了提高其性能,我们将该算法扩展为一种并行算法,通过Hadoop MapReduce框架并行复制数据。所提出的MapReduce并行组合算法在从HapMap数据集中获取的真实世界数据上表现良好;计算效率的提高与所使用的处理器数量成正比。