Ceballos-Picot Irène, Le Dantec Aurélia, Brassier Anaïs, Jaïs Jean-Philippe, Ledroit Morgan, Cahu Julie, Ea Hang-Korng, Daignan-Fornier Bertrand, Pinson Benoît
Laboratoire de Biochimie métabolomique et protéomique, Hôpital Necker-Enfants Malades, AP-HP, 149 rue de Sèvres, Paris, 75015, France.
Université Paris Descartes Sorbonne Paris Cité, 15 rue de l'Ecole de Médecine, Paris, 75006, France.
Orphanet J Rare Dis. 2015 Jan 23;10:7. doi: 10.1186/s13023-014-0219-0.
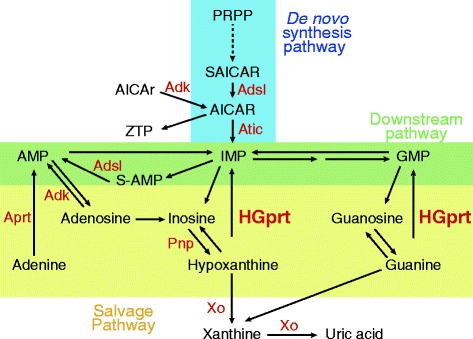
Lesch-Nyhan disease is a rare X-linked neurodevelopemental metabolic disorder caused by a wide variety of mutations in the HPRT1 gene leading to a deficiency of the purine recycling enzyme hypoxanthine-guanine phosphoribosyltransferase (HGprt). The residual HGprt activity correlates with the various phenotypes of Lesch-Nyhan (LN) patients and in particular with the different degree of neurobehavioral disturbances. The prevalence of this disease is considered to be underestimated due to large heterogeneity of its clinical symptoms and the difficulty of diagnosing of the less severe forms of the disease. We therefore searched for metabolic changes that would facilitate an early diagnosis and give potential clues on the disease pathogenesis and potential therapeutic approaches.
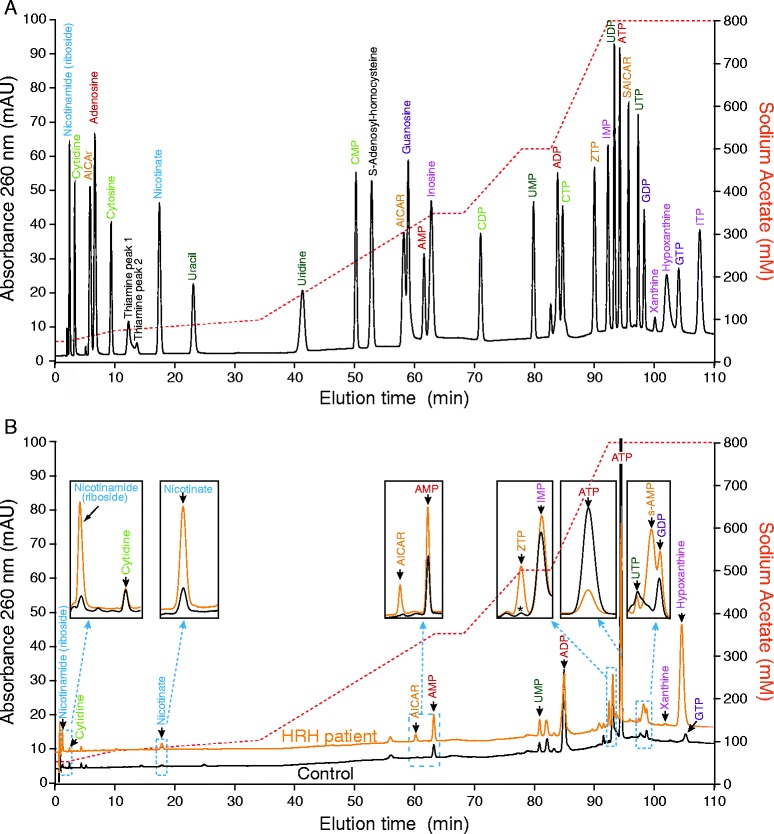
Lesch-Nyhan patients were diagnosed using HGprt enzymatic assay in red blood cells and identification of the causal HPRT1 gene mutations. These patients were subsequently classified into the three main phenotypic subgroups ranging from patients with only hyperuricemia to individuals presenting motor dysfunction, cognitive disability and self-injurious behavior. Metabolites from the three classes of patients were analyzed and quantified by High Performance Ionic Chromatography and biomarkers of HGprt deficiency were then validated by statistical analyses.
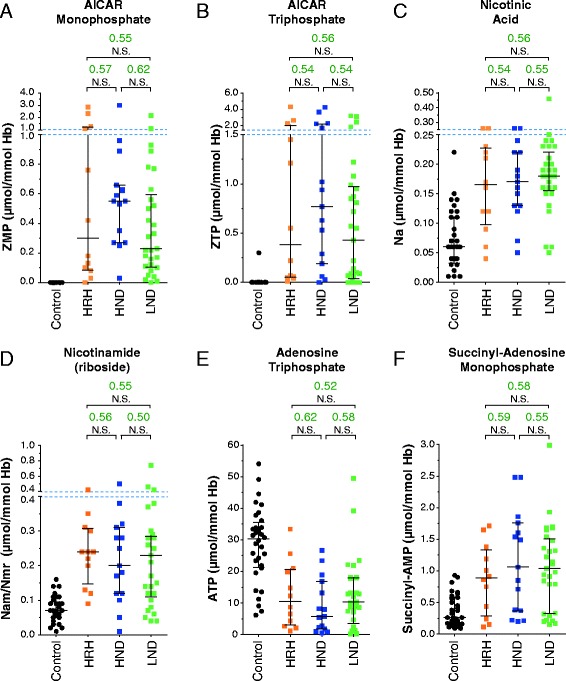
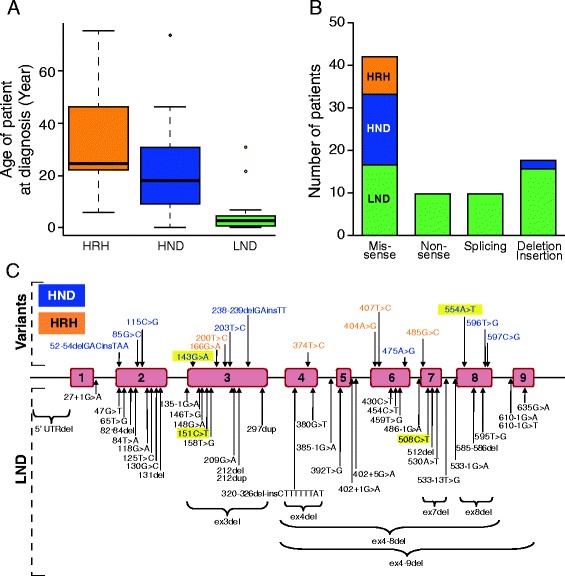
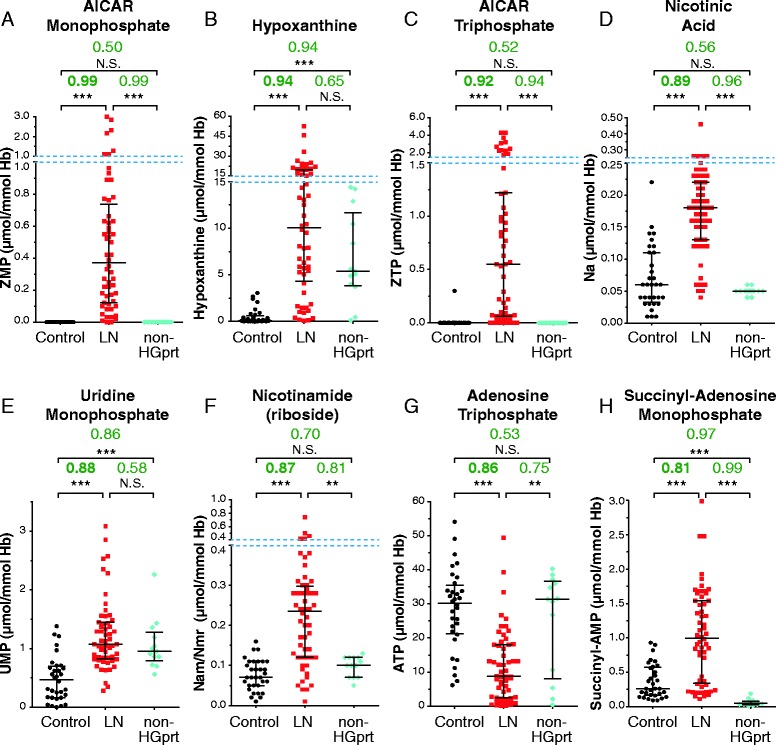
A cohort of 139 patients, from 112 families, diagnosed using HGprt enzymatic assay in red blood cells, was studied. 98 displayed LN full phenotype (86 families) and 41 (26 families) had attenuated clinical phenotypes. Genotype/phenotype correlations show that LN full phenotype was correlated to genetic alterations resulting in null enzyme function, while variant phenotypes are often associated with missense mutations allowing some residual HGprt activity. Analysis of metabolites extracted from red blood cells from 56 LN patients revealed strong variations specific to HGprt deficiency for six metabolites (AICAR mono- and tri-phosphate, nicotinamide, nicotinic acid, ATP and Succinyl-AMP) as compared to controls including hyperuricemic patients without HGprt deficiency.
A highly significant correlation between six metabolites and the HGprt deficiency was established, each of them providing an easily measurable marker of the disease. Their combination strongly increases the probability of an early and reliable diagnosis for HGprt deficiency.
莱施-奈恩病是一种罕见的X连锁神经发育性代谢紊乱疾病,由HPRT1基因的多种突变引起,导致嘌呤回收酶次黄嘌呤-鸟嘌呤磷酸核糖转移酶(HGprt)缺乏。残余的HGprt活性与莱施-奈恩(LN)患者的各种表型相关,特别是与不同程度的神经行为障碍相关。由于该疾病临床症状的高度异质性以及诊断较轻形式疾病的困难,其患病率被认为被低估。因此,我们寻找有助于早期诊断并为疾病发病机制和潜在治疗方法提供潜在线索的代谢变化。
通过红细胞中HGprt酶活性测定和致病HPRT1基因突变鉴定来诊断莱施-奈恩病患者。这些患者随后被分为三个主要表型亚组,从仅患有高尿酸血症的患者到出现运动功能障碍、认知残疾和自伤行为的个体。通过高效离子色谱法分析和定量这三类患者的代谢物,然后通过统计分析验证HGprt缺乏的生物标志物。
研究了一组139名患者,来自112个家庭,通过红细胞中HGprt酶活性测定进行诊断。98名表现出LN完全表型(86个家庭),41名(26个家庭)具有临床表型减弱。基因型/表型相关性表明,LN完全表型与导致酶功能缺失的基因改变相关,而变异表型通常与允许一些残余HGprt活性的错义突变相关。对56名LN患者红细胞中提取的代谢物分析显示,与包括无HGprt缺乏的高尿酸血症患者在内的对照组相比,六种代谢物(一磷酸和三磷酸氨基咪唑甲酰胺、烟酰胺、烟酸酯、ATP和琥珀酰-AMP)存在特定于HGprt缺乏的强烈变化。
六种代谢物与HGprt缺乏之间建立了高度显著的相关性,每种代谢物都提供了一种易于测量的疾病标志物。它们的组合大大增加了HGprt缺乏早期和可靠诊断的可能性。