Klein S R, Piya S, Lu Z, Xia Y, Alonso M M, White E J, Wei J, Gomez-Manzano C, Jiang H, Fueyo J
Department of Neuro-Oncology, Brain Tumor Center, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Cancer Biology Program, The University of Texas Graduate School of Biomedical Sciences at Houston, Houston, TX, USA.
Oncogene. 2015 Oct 8;34(41):5295-301. doi: 10.1038/onc.2014.452. Epub 2015 Jan 26.
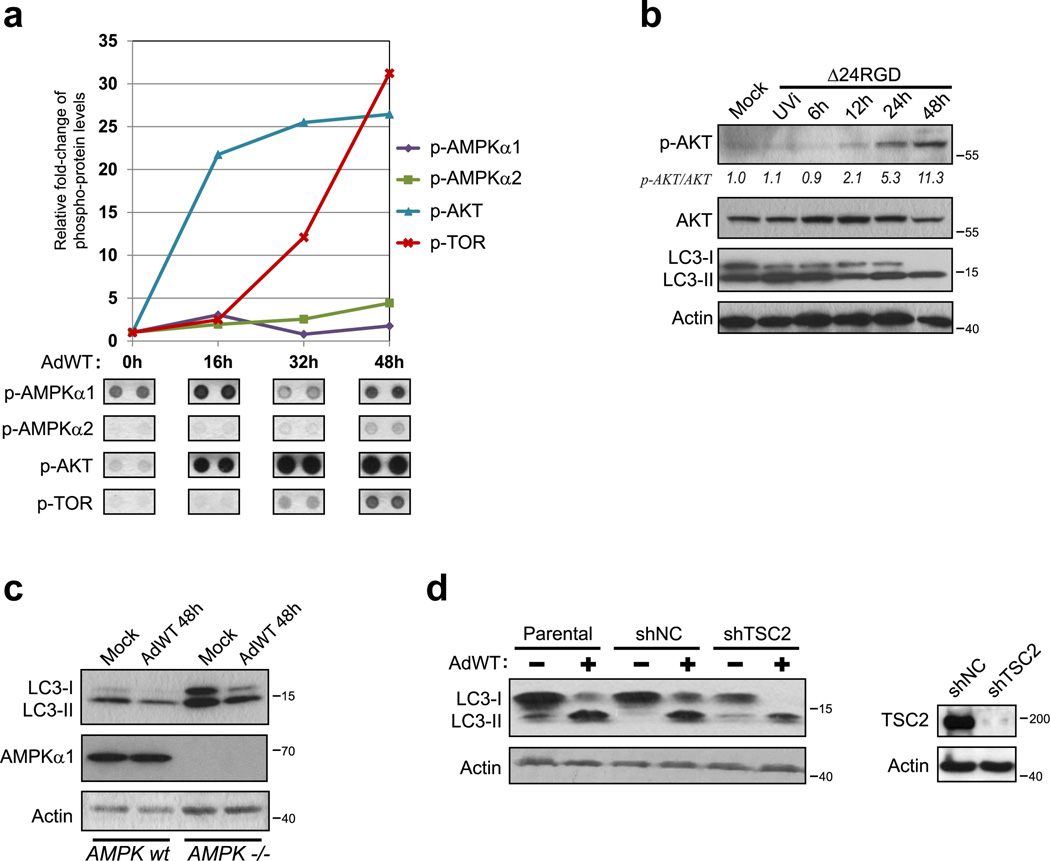
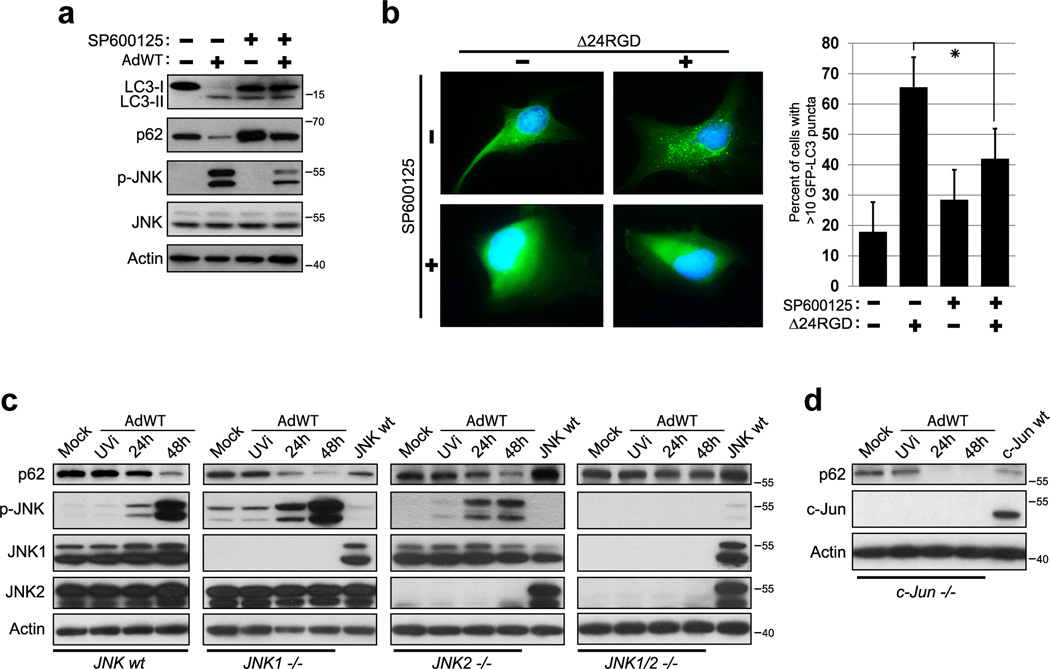
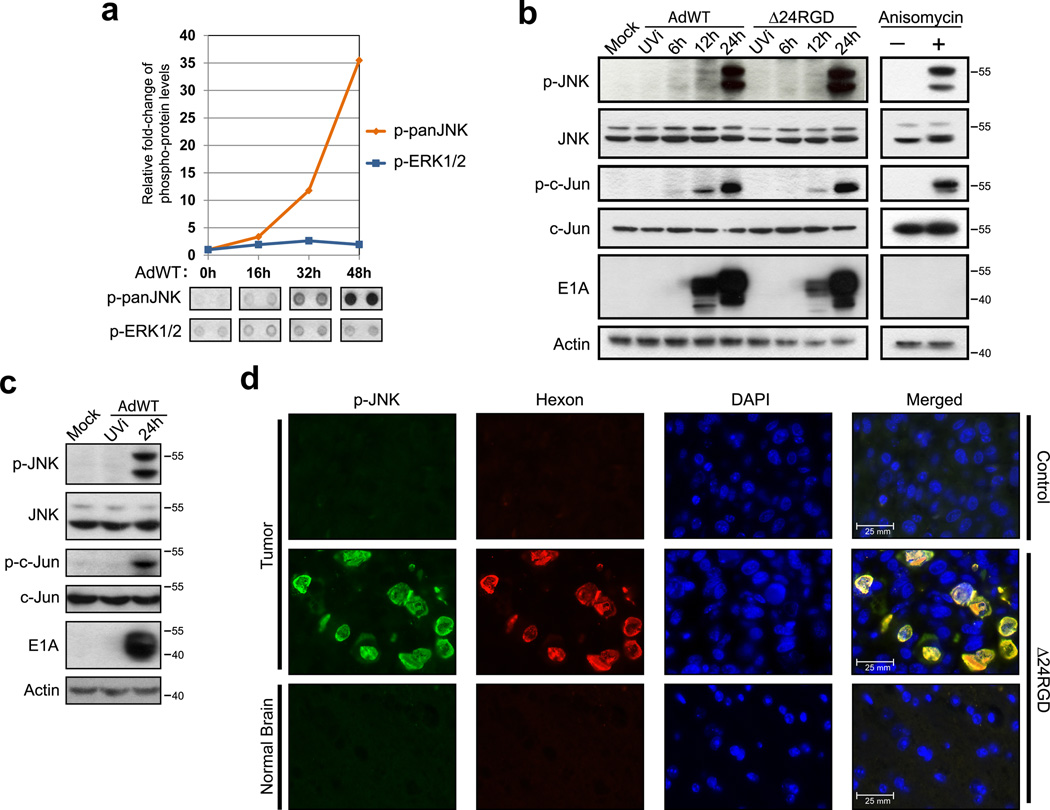
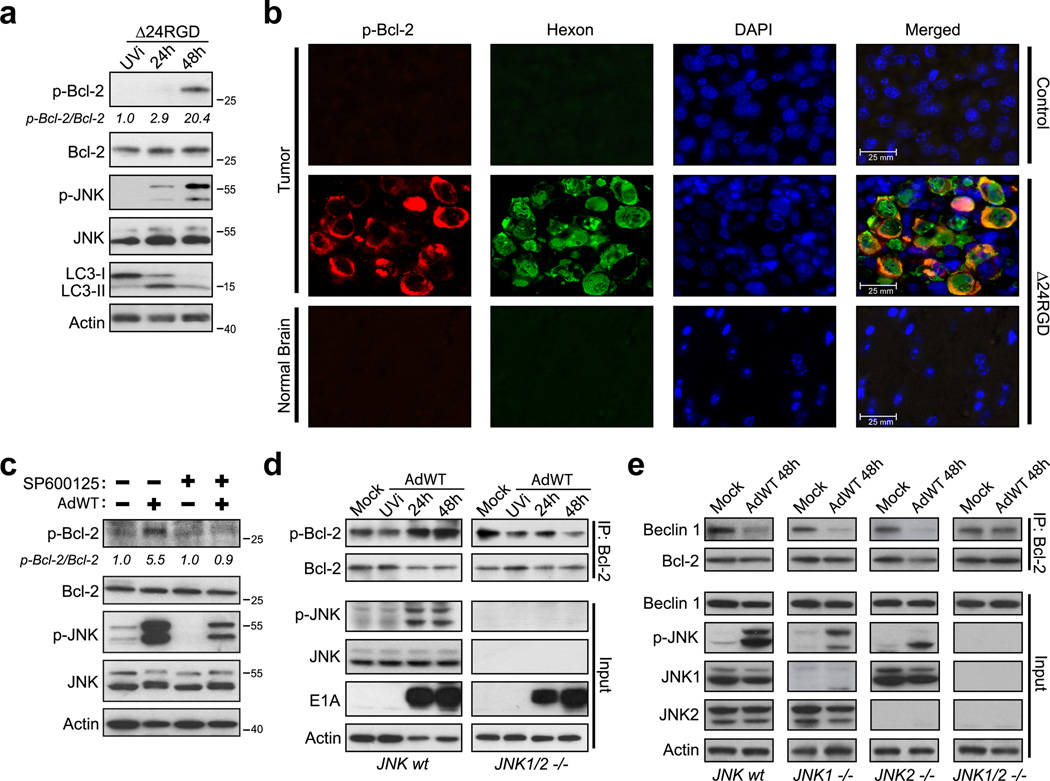
Oncolytic adenoviruses, such as Delta-24-RGD (Δ24RGD), are replication-competent viruses that are genetically engineered to induce selective cancer cell lysis. In cancer cells, Δ24RGD induces massive autophagy, which is required for efficient cell lysis and adenoviral spread. Understanding the cellular mechanisms underlying the regulation of autophagy in cells treated with oncolytic adenoviruses may provide new avenues to improve the therapeutic effect. In this work, we showed that cancer cells infected with Δ24RGDundergo autophagy despite the concurrent activation of the AKT/mTOR pathway. Moreover, adenovirus replication induced sustained activation of JNK proteins in vitro. ERK1/2 phosphorylation remained unchanged during adenoviral infection, suggesting specificity of JNK activation. Using genetic ablation and pharmacological inactivation of JNK, we unequivocally demonstrated that cells infected with Δ24RGD required JNK activation. Thus, genetic co-ablation of JNK1 and JNK2 genes or inhibition of JNK kinase function rendered Δ24RGD-treated cells resistant to autophagy. Accordingly, JNK activation induced phosphorylation of Bcl-2 and prevented the formation of Bcl-2/Beclin 1 autophagy suppressor complexes. Using an orthotopic model of human glioma xenograft, we showed that treatment with Δ24RGD induced phosphorylation and nuclear translocation of JNK, as well as phosphorylation of Bcl-2. Collectively, our data identified JNK proteins as an essential mechanistic link between Δ24RGD infection and autophagy in cancer cells. Activation of JNK without inactivation of the AKT/mTOR pathway constitutes a distinct molecular signature of autophagy regulation that differentiates Δ24RGD adenovirus from the mechanism used by other oncolytic viruses to induce autophagy and provides a new rationale for the combination of oncolytic viruses and chemotherapy.
溶瘤腺病毒,如Delta-24-RGD(Δ24RGD),是具有复制能力的病毒,经过基因工程改造以诱导选择性癌细胞裂解。在癌细胞中,Δ24RGD诱导大量自噬,这是有效细胞裂解和腺病毒传播所必需的。了解溶瘤腺病毒处理的细胞中自噬调节的细胞机制可能为提高治疗效果提供新途径。在这项工作中,我们表明感染Δ24RGD的癌细胞尽管同时激活了AKT/mTOR途径,但仍会发生自噬。此外,腺病毒复制在体外诱导JNK蛋白持续激活。在腺病毒感染期间ERK1/2磷酸化保持不变,表明JNK激活具有特异性。使用JNK的基因敲除和药理学失活,我们明确证明感染Δ24RGD的细胞需要JNK激活。因此,JNK1和JNK2基因的基因共敲除或JNK激酶功能的抑制使Δ24RGD处理的细胞对自噬产生抗性。相应地,JNK激活诱导Bcl-2磷酸化并阻止Bcl-2/Beclin 1自噬抑制复合物的形成。使用人胶质瘤异种移植的原位模型,我们表明用Δ24RGD处理可诱导JNK磷酸化和核转位以及Bcl-2磷酸化。总体而言,我们的数据确定JNK蛋白是Δ24RGD感染与癌细胞自噬之间的重要机制联系。JNK的激活而不使AKT/mTOR途径失活构成了自噬调节的独特分子特征,这使Δ24RGD腺病毒与其他溶瘤病毒诱导自噬的机制不同,并为溶瘤病毒与化疗联合提供了新的理论依据。