Floudas Charalampos S, Um Nara, Kamboh M Ilyas, Barmada Michael M, Visweswaran Shyam
Department of Biomedical Informatics, University of Pittsburgh, 5607 Baum Boulevard, Pittsburgh, PA 15206 USA.
Department of Human Genetics, University of Pittsburgh, 130 De Soto Street, Pittsburgh, PA 15261 USA.
BioData Min. 2014 Dec 19;7(1):35. doi: 10.1186/s13040-014-0035-z. eCollection 2014.
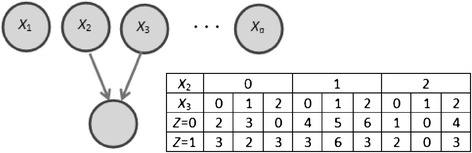
Identifying genetic interactions in data obtained from genome-wide association studies (GWASs) can help in understanding the genetic basis of complex diseases. The large number of single nucleotide polymorphisms (SNPs) in GWASs however makes the identification of genetic interactions computationally challenging. We developed the Bayesian Combinatorial Method (BCM) that can identify pairs of SNPs that in combination have high statistical association with disease.
We applied BCM to two late-onset Alzheimer's disease (LOAD) GWAS datasets to identify SNPs that interact with known Alzheimer associated SNPs. We also compared BCM with logistic regression that is implemented in PLINK. Gene Ontology analysis of genes from the top 200 dataset SNPs for both GWAS datasets showed overrepresentation of LOAD-related terms. Four genes were common to both datasets: APOE and APOC1, which have well established associations with LOAD, and CAMK1D and FBXL13, not previously linked to LOAD but having evidence of involvement in LOAD. Supporting evidence was also found for additional genes from the top 30 dataset SNPs.
BCM performed well in identifying several SNPs having evidence of involvement in the pathogenesis of LOAD that would not have been identified by univariate analysis due to small main effect. These results provide support for applying BCM to identify potential genetic variants such as SNPs from high dimensional GWAS datasets.
在全基因组关联研究(GWAS)获得的数据中识别基因相互作用有助于理解复杂疾病的遗传基础。然而,GWAS中大量的单核苷酸多态性(SNP)使得基因相互作用的识别在计算上具有挑战性。我们开发了贝叶斯组合方法(BCM),该方法可以识别组合起来与疾病具有高度统计关联的SNP对。
我们将BCM应用于两个迟发性阿尔茨海默病(LOAD)GWAS数据集,以识别与已知阿尔茨海默病相关SNP相互作用的SNP。我们还将BCM与PLINK中实现的逻辑回归进行了比较。对两个GWAS数据集的前200个数据集中SNP的基因进行基因本体分析,结果显示与LOAD相关的术语过度富集。两个数据集共有四个基因:与LOAD有明确关联的APOE和APOC1,以及之前未与LOAD相关联但有证据表明参与LOAD的CAMK1D和FBXL13。在前30个数据集中SNP的其他基因也发现了支持证据。
BCM在识别几个有证据表明参与LOAD发病机制的SNP方面表现良好,这些SNP由于主效应较小,单变量分析无法识别。这些结果为应用BCM识别潜在的遗传变异(如来自高维GWAS数据集的SNP)提供了支持。