Jiang Xia, Neapolitan Richard E
Department of Biomedical Informatics, University of Pittsburgh, Pittsburgh, Pennsylvania, United States of America.
Genet Epidemiol. 2015 Mar;39(3):173-84. doi: 10.1002/gepi.21889. Epub 2015 Feb 12.
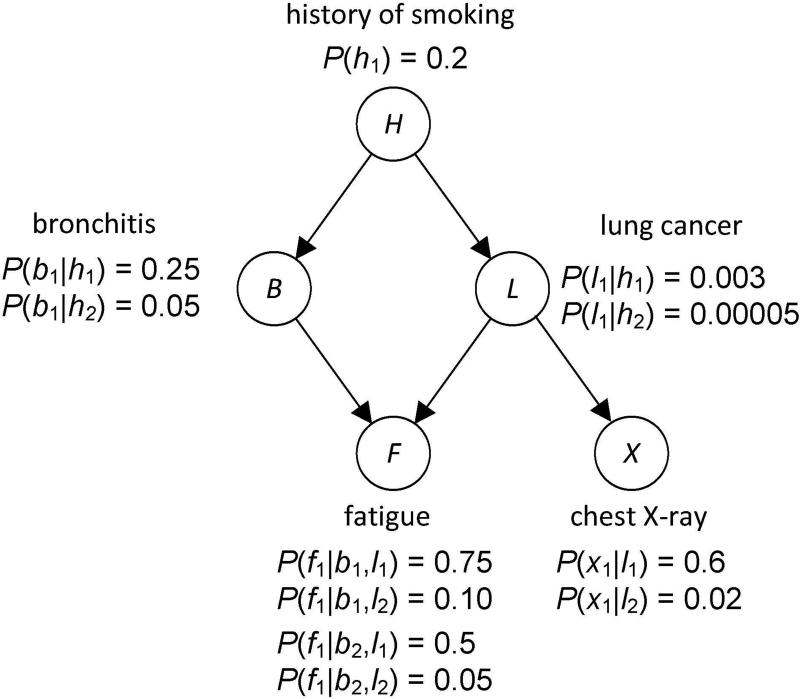
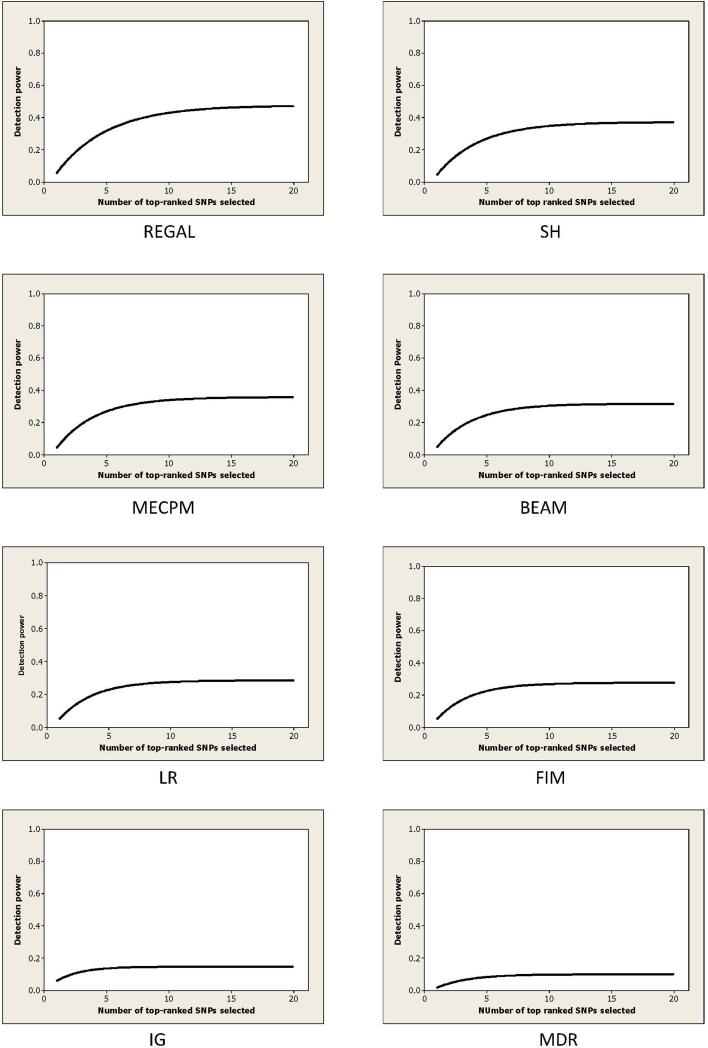
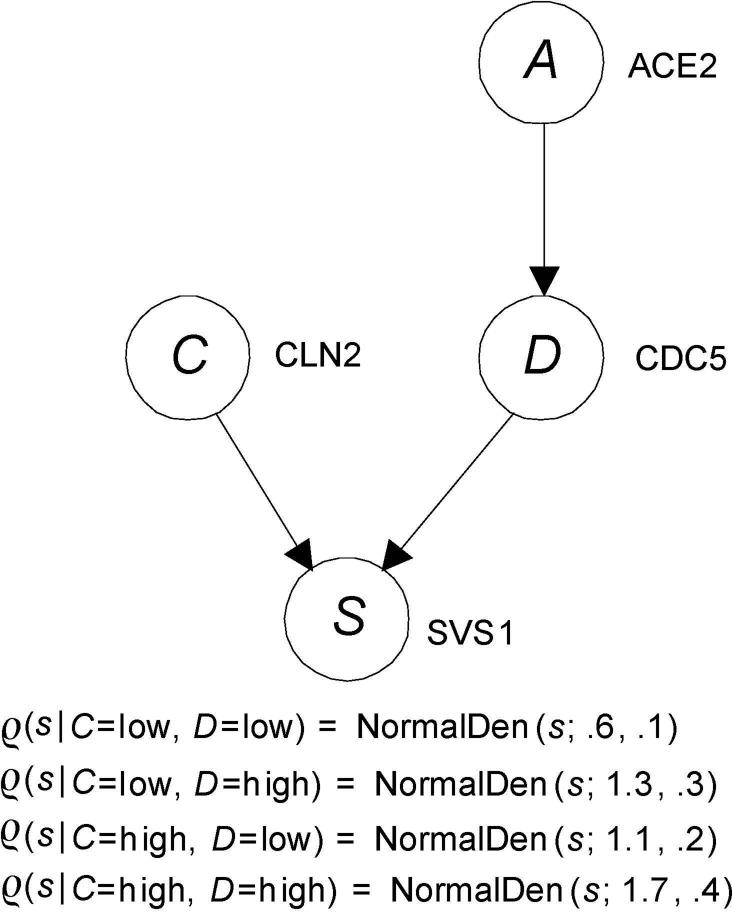
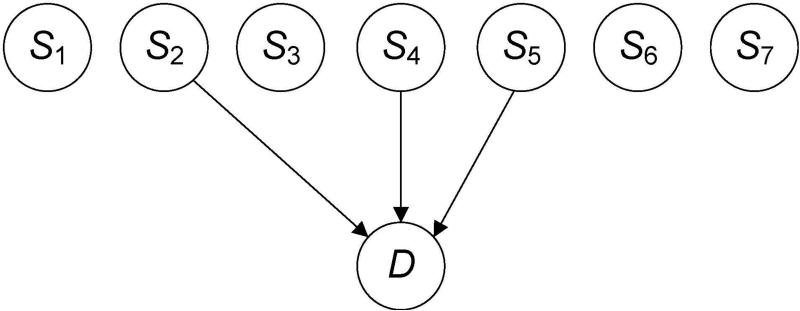
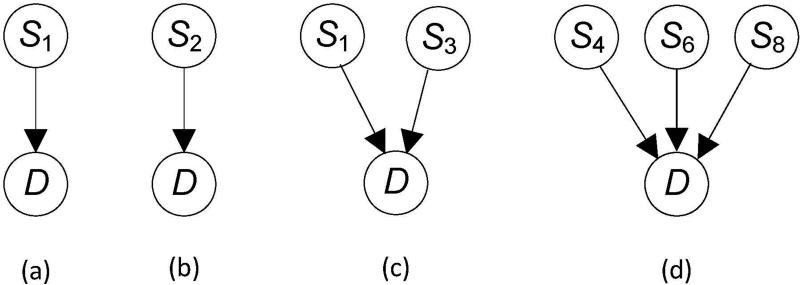
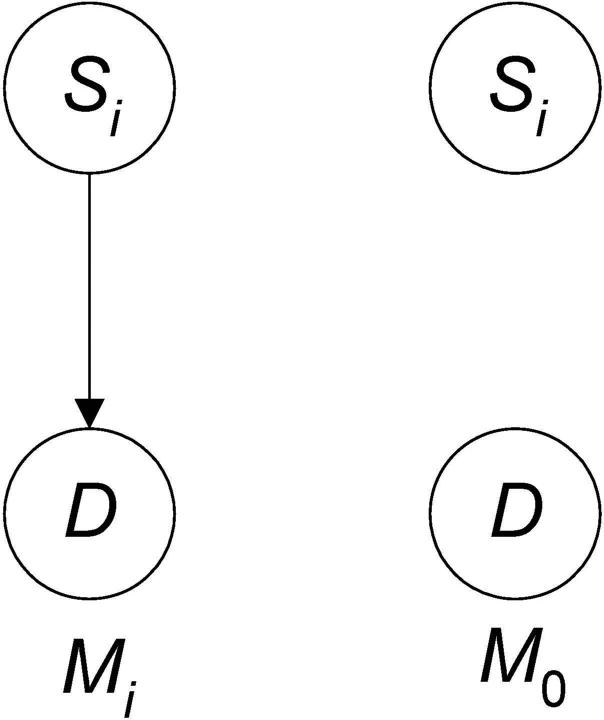
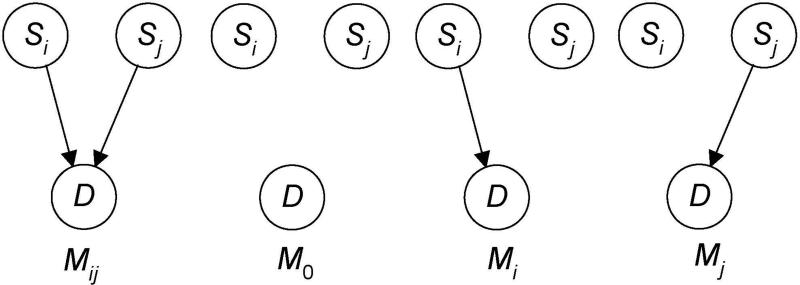
Single nucleotide polymorphism (SNP) high-dimensional datasets are available from Genome Wide Association Studies (GWAS). Such data provide researchers opportunities to investigate the complex genetic basis of diseases. Much of genetic risk might be due to undiscovered epistatic interactions, which are interactions in which combination of several genes affect disease. Research aimed at discovering interacting SNPs from GWAS datasets proceeded in two directions. First, tools were developed to evaluate candidate interactions. Second, algorithms were developed to search over the space of candidate interactions. Another problem when learning interacting SNPs, which has not received much attention, is evaluating how likely it is that the learned SNPs are associated with the disease. A complete system should provide this information as well. We develop such a system. Our system, called LEAP, includes a new heuristic search algorithm for learning interacting SNPs, and a Bayesian network based algorithm for computing the probability of their association. We evaluated the performance of LEAP using 100 1,000-SNP simulated datasets, each of which contains 15 SNPs involved in interactions. When learning interacting SNPs from these datasets, LEAP outperformed seven others methods. Furthermore, only SNPs involved in interactions were found to be probable. We also used LEAP to analyze real Alzheimer's disease and breast cancer GWAS datasets. We obtained interesting and new results from the Alzheimer's dataset, but limited results from the breast cancer dataset. We conclude that our results support that LEAP is a useful tool for extracting candidate interacting SNPs from high-dimensional datasets and determining their probability.
单核苷酸多态性(SNP)高维数据集可从全基因组关联研究(GWAS)中获取。此类数据为研究人员提供了调查疾病复杂遗传基础的机会。许多遗传风险可能归因于尚未发现的上位性相互作用,即几个基因的组合影响疾病的相互作用。旨在从GWAS数据集中发现相互作用SNP的研究朝着两个方向进行。首先,开发了工具来评估候选相互作用。其次,开发了算法来在候选相互作用空间中进行搜索。在学习相互作用SNP时,另一个未受到太多关注的问题是评估所学习的SNP与疾病相关的可能性。一个完整的系统也应该提供此信息。我们开发了这样一个系统。我们的系统称为LEAP,包括一种用于学习相互作用SNP的新启发式搜索算法,以及一种基于贝叶斯网络的算法来计算它们关联的概率。我们使用100个1000-SNP模拟数据集评估了LEAP的性能,每个数据集包含15个参与相互作用的SNP。当从这些数据集中学习相互作用SNP时,LEAP的表现优于其他七种方法。此外,仅发现参与相互作用的SNP是可能的。我们还使用LEAP分析了真实的阿尔茨海默病和乳腺癌GWAS数据集。我们从阿尔茨海默病数据集中获得了有趣的新结果,但从乳腺癌数据集中获得的结果有限。我们得出结论,我们的结果支持LEAP是从高维数据集中提取候选相互作用SNP并确定其概率的有用工具。