Jeong Hyeon-Uk, Kwon Mihwa, Lee Yongnam, Yoo Ji Seok, Shin Dae Hee, Song Im-Sook, Lee Hye Suk
College of Pharmacy, The Catholic University of Korea, Bucheon 420-743, Korea.
College of Pharmacy and Research Institute of Pharmaceutical Sciences, Kyungpook National University, Daegu 702-701, Korea.
Drug Des Devel Ther. 2015 Jan 22;9:643-53. doi: 10.2147/DDDT.S75400. eCollection 2015.
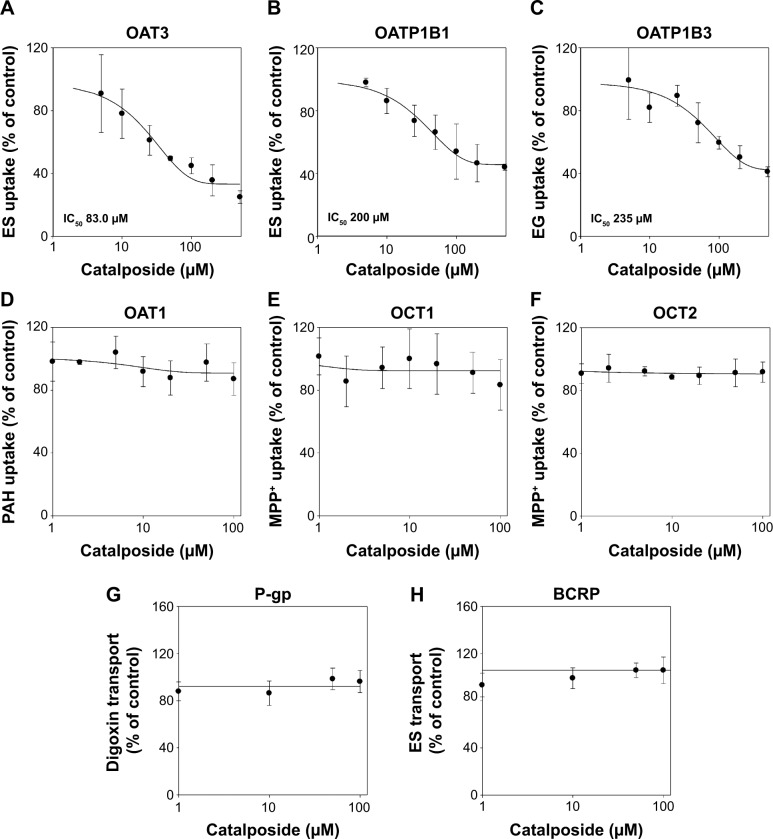
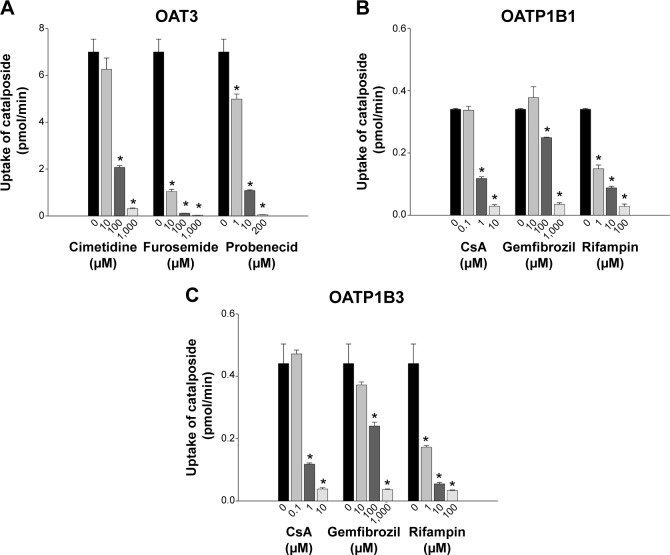
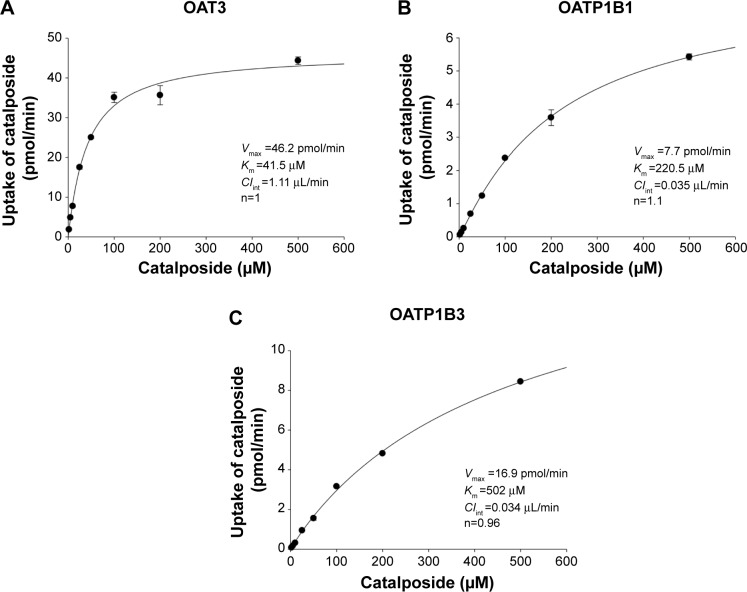
We investigated the in vitro transport characteristics of catalposide in HEK293 cells overexpressing organic anion transporter 1 (OAT1), OAT3, organic anion transporting polypeptide 1B1 (OATP1B1), OATP1B3, organic cation transporter 1 (OCT1), OCT2, P-glycoprotein (P-gp), and breast cancer resistance protein (BCRP). The transport mechanism of catalposide was investigated in HEK293 and LLC-PK1 cells overexpressing the relevant transporters. The uptake of catalposide was 319-, 13.6-, and 9.3-fold greater in HEK293 cells overexpressing OAT3, OATP1B1, and OATP1B3 transporters, respectively, than in HEK293 control cells. The increased uptake of catalposide via the OAT3, OATP1B1, and OATP1B3 transporters was decreased to basal levels in the presence of representative inhibitors such as probenecid, furosemide, and cimetidine (for OAT3) and cyclosporin A, gemfibrozil, and rifampin (for OATP1B1 and OATP1B3). The concentration-dependent OAT3-mediated uptake of catalposide revealed the following kinetic parameters: Michaelis constant (K m) =41.5 μM, maximum uptake rate (V max) =46.2 pmol/minute, and intrinsic clearance (CL int) =1.11 μL/minute. OATP1B1- and OATP1B3-mediated catalposide uptake also showed concentration dependency, with low CL int values of 0.035 and 0.034 μL/minute, respectively. However, the OCT1, OCT2, OAT1, P-gp, and BCRP transporters were apparently not involved in the uptake of catalposide into cells. In addition, catalposide inhibited the transport activities of OAT3, OATP1B1, and OATP1B3 with half-maximal inhibitory concentration values of 83, 200, and 235 μM, respectively. However, catalposide did not significantly inhibit the transport activities of OCT1, OCT2, OAT1, P-gp, or BCRP. In conclusion, OAT3, OATP1B1, and OATP1B3 are major transporters that may regulate the pharmacokinetic properties and may cause herb-drug interactions of catalposide, although their clinical relevance awaits further evaluation.
我们研究了梓醇在过表达有机阴离子转运体1(OAT1)、OAT3、有机阴离子转运多肽1B1(OATP1B1)、OATP1B3、有机阳离子转运体1(OCT1)、OCT2、P-糖蛋白(P-gp)和乳腺癌耐药蛋白(BCRP)的HEK293细胞中的体外转运特性。在过表达相关转运体的HEK293和LLC-PK1细胞中研究了梓醇的转运机制。过表达OAT3、OATP1B1和OATP1B3转运体的HEK293细胞对梓醇的摄取分别比HEK293对照细胞高319倍、13.6倍和9.3倍。在存在丙磺舒、呋塞米和西咪替丁(针对OAT3)以及环孢素A、吉非贝齐和利福平(针对OATP1B1和OATP1B3)等代表性抑制剂的情况下,通过OAT3、OATP1B1和OATP1B3转运体增加的梓醇摄取降至基础水平。梓醇的浓度依赖性OAT3介导的摄取显示出以下动力学参数:米氏常数(Km)=41.5μM,最大摄取速率(Vmax)=46.2 pmol/分钟,内在清除率(CLint)=1.11μL/分钟。OATP1B1和OATP1B3介导的梓醇摄取也显示出浓度依赖性,CLint值分别较低,为0.035和0.034μL/分钟。然而,OCT1、OCT2、OAT1、P-gp和BCRP转运体显然不参与梓醇进入细胞的摄取。此外,梓醇抑制OAT3、OATP1B1和OATP1B3的转运活性,半数最大抑制浓度值分别为83、200和235μM。然而,梓醇并未显著抑制OCT1、OCT2、OAT1、P-gp或BCRP的转运活性。总之,OAT3、OATP1B1和OATP1B3是主要的转运体,可能调节梓醇的药代动力学性质并可能导致其与药物的相互作用,尽管它们的临床相关性有待进一步评估。