Wu Qian, Won Kyoung-Jae, Li Hongzhe
Department of Biostatistics and Genetics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Cancer Inform. 2015 Jan 27;14(Suppl 1):11-22. doi: 10.4137/CIN.S13972. eCollection 2015.
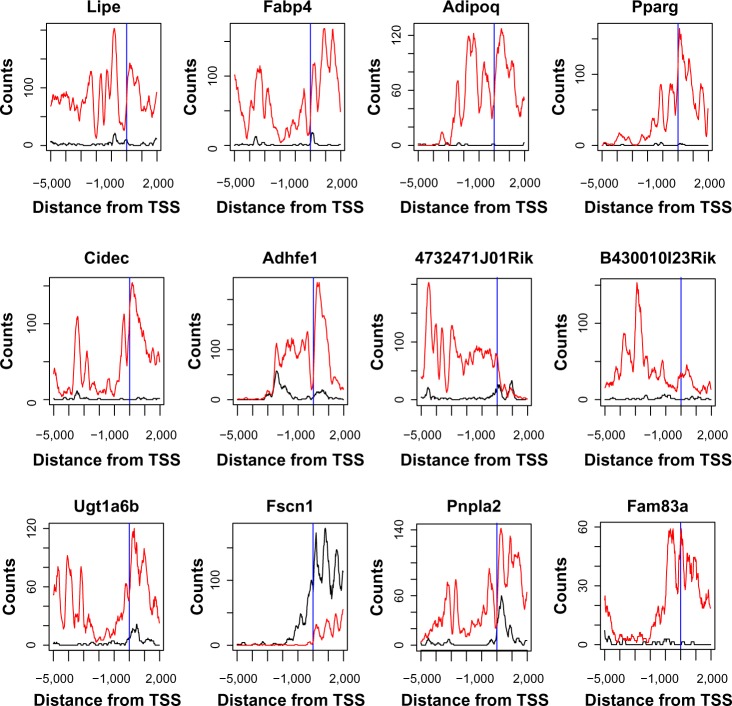
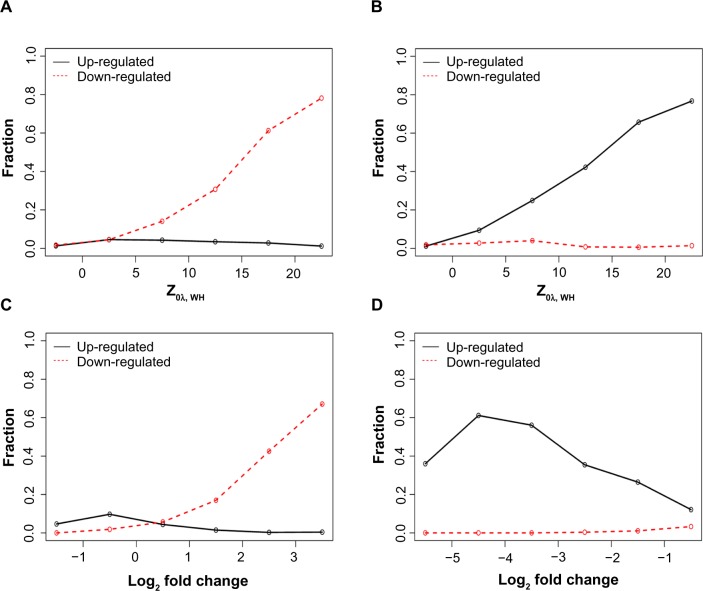
Chromatin immunoprecipitation sequencing (ChIP-seq) is a powerful method for analyzing protein interactions with DNA. It can be applied to identify the binding sites of transcription factors (TFs) and genomic landscape of histone modification marks (HMs). Previous research has largely focused on developing peak-calling procedures to detect the binding sites for TFs. However, these procedures may fail when applied to ChIP-seq data of HMs, which have diffuse signals and multiple local peaks. In addition, it is important to identify genes with differential histone enrichment regions between two experimental conditions, such as different cellular states or different time points. Parametric methods based on Poisson/negative binomial distribution have been proposed to address this differential enrichment problem and most of these methods require biological replications. However, many ChIP-seq data usually have a few or even no replicates. We propose a nonparametric method to identify the genes with differential histone enrichment regions even without replicates. Our method is based on nonparametric hypothesis testing and kernel smoothing in order to capture the spatial differences in histone-enriched profiles. We demonstrate the method using ChIP-seq data on a comparative epigenomic profiling of adipogenesis of murine adipose stromal cells and the Encyclopedia of DNA Elements (ENCODE) ChIP-seq data. Our method identifies many genes with differential H3K27ac histone enrichment profiles at gene promoter regions between proliferating preadipocytes and mature adipocytes in murine 3T3-L1 cells. The test statistics also correlate with the gene expression changes well and are predictive to gene expression changes, indicating that the identified differentially enriched regions are indeed biologically meaningful.
染色质免疫沉淀测序(ChIP-seq)是一种分析蛋白质与DNA相互作用的强大方法。它可用于识别转录因子(TFs)的结合位点以及组蛋白修饰标记(HMs)的基因组图谱。先前的研究主要集中在开发峰检测程序以检测TFs的结合位点。然而,当应用于具有弥散信号和多个局部峰的HMs的ChIP-seq数据时,这些程序可能会失败。此外,识别在两种实验条件(如不同细胞状态或不同时间点)之间具有差异组蛋白富集区域的基因也很重要。已经提出了基于泊松/负二项分布的参数方法来解决这种差异富集问题,并且这些方法中的大多数都需要生物学重复。然而,许多ChIP-seq数据通常只有很少甚至没有重复。我们提出了一种非参数方法,即使在没有重复的情况下也能识别具有差异组蛋白富集区域的基因。我们的方法基于非参数假设检验和核平滑,以捕获组蛋白富集图谱中的空间差异。我们使用小鼠脂肪基质细胞脂肪生成的比较表观基因组图谱的ChIP-seq数据和DNA元件百科全书(ENCODE)ChIP-seq数据来演示该方法。我们的方法在小鼠3T3-L1细胞中增殖前脂肪细胞和成熟脂肪细胞之间的基因启动子区域识别出许多具有差异H3K27ac组蛋白富集图谱的基因。检验统计量也与基因表达变化高度相关,并且能够预测基因表达变化,这表明所识别的差异富集区域确实具有生物学意义。