Fukasawa Yoshinori, Tsuji Junko, Fu Szu-Chin, Tomii Kentaro, Horton Paul, Imai Kenichiro
From the ‡Department of Computational Biology, Graduate School of Frontier Sciences, The University Tokyo, 5-1-5, Kashiwanoha, Kashiwa, Chiba, 277-8561, Japan;
From the ‡Department of Computational Biology, Graduate School of Frontier Sciences, The University Tokyo, 5-1-5, Kashiwanoha, Kashiwa, Chiba, 277-8561, Japan; §Computational Biology Research Center, National Institute of Advanced Industrial Science and Technology (AIST), 2-4-7 Aomi, Koto-ku, Tokyo 135-0064, Japan.
Mol Cell Proteomics. 2015 Apr;14(4):1113-26. doi: 10.1074/mcp.M114.043083. Epub 2015 Feb 10.
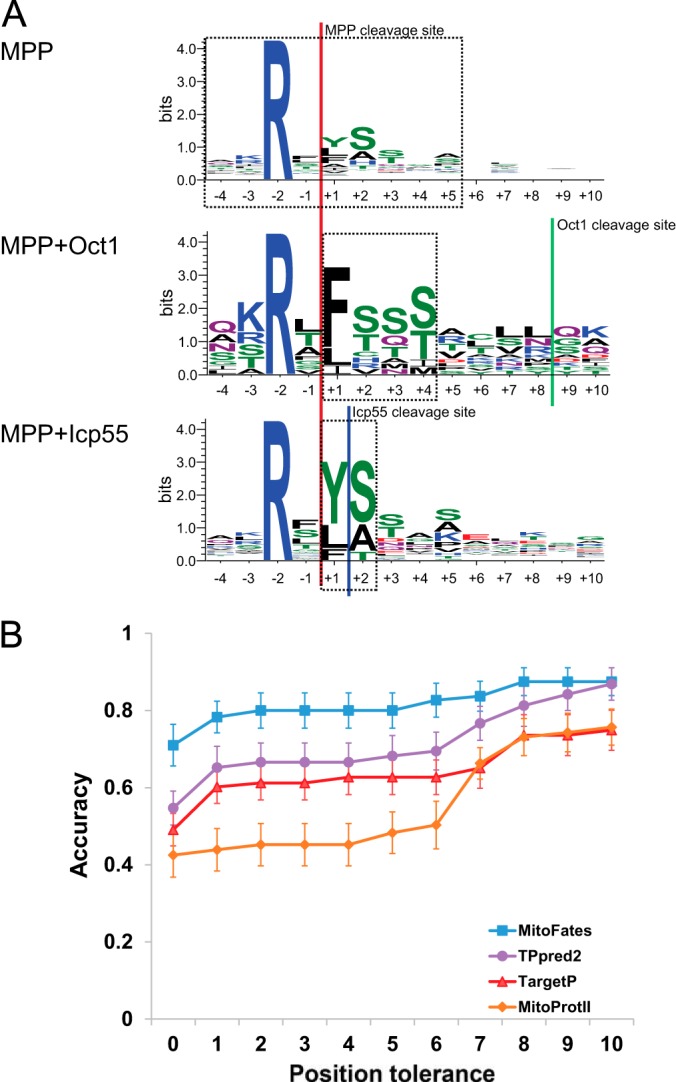
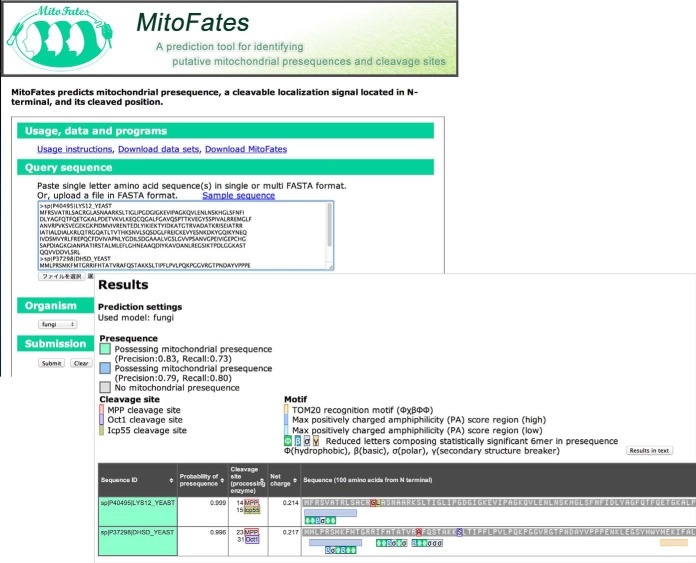
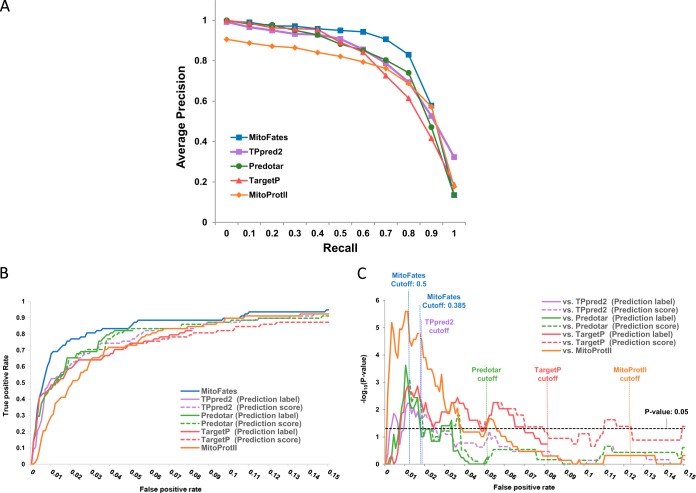
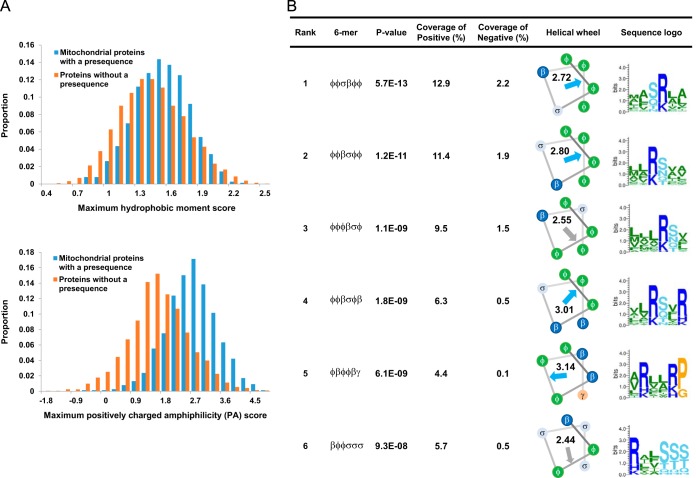
Mitochondria provide numerous essential functions for cells and their dysfunction leads to a variety of diseases. Thus, obtaining a complete mitochondrial proteome should be a crucial step toward understanding the roles of mitochondria. Many mitochondrial proteins have been identified experimentally but a complete list is not yet available. To fill this gap, methods to computationally predict mitochondrial proteins from amino acid sequence have been developed and are widely used, but unfortunately, their accuracy is far from perfect. Here we describe MitoFates, an improved prediction method for cleavable N-terminal mitochondrial targeting signals (presequences) and their cleavage sites. MitoFates introduces novel sequence features including positively charged amphiphilicity, presequence motifs, and position weight matrices modeling the presequence cleavage sites. These features are combined with classical ones such as amino acid composition and physico-chemical properties as input to a standard support vector machine classifier. On independent test data, MitoFates attains better performance than existing predictors in both detection of presequences and in predicting their cleavage sites. We used MitoFates to look for undiscovered mitochondrial proteins from 42,217 human proteins (including isoforms such as alternative splicing or translation initiation variants). MitoFates predicts 1167 genes to have at least one isoform with a presequence. Five-hundred and eighty of these genes were not annotated as mitochondrial in either UniProt or Gene Ontology. Interestingly, these include candidate regulators of parkin translocation to damaged mitochondria, and also many genes with known disease mutations, suggesting that careful investigation of MitoFates predictions may be helpful in elucidating the role of mitochondria in health and disease. MitoFates is open source with a convenient web server publicly available.
线粒体为细胞提供众多重要功能,其功能障碍会导致多种疾病。因此,获取完整的线粒体蛋白质组应是理解线粒体作用的关键一步。许多线粒体蛋白质已通过实验鉴定出来,但完整列表尚未可得。为填补这一空白,已开发出从氨基酸序列计算预测线粒体蛋白质的方法并被广泛使用,但遗憾的是,其准确性远非完美。在此,我们描述了MitoFates,一种用于可裂解N端线粒体靶向信号(前序列)及其裂解位点的改进预测方法。MitoFates引入了新的序列特征,包括带正电荷的两亲性、前序列基序以及对前序列裂解位点进行建模的位置权重矩阵。这些特征与诸如氨基酸组成和物理化学性质等经典特征相结合,作为标准支持向量机分类器的输入。在独立测试数据上,MitoFates在检测前序列及其裂解位点的预测方面均比现有预测器表现更好。我们使用MitoFates从42217个人类蛋白质(包括可变剪接或翻译起始变体等同型)中寻找未发现的线粒体蛋白质。MitoFates预测1167个基因至少有一个具有前序列的异构体。其中580个基因在UniProt或基因本体中均未被注释为线粒体基因。有趣的是,这些基因包括帕金蛋白向受损线粒体转位的候选调节因子,以及许多具有已知疾病突变的基因,这表明仔细研究MitoFates的预测结果可能有助于阐明线粒体在健康和疾病中的作用。MitoFates是开源的,并有一个方便的公共网络服务器可供使用。