Esling Philippe, Lejzerowicz Franck, Pawlowski Jan
Department of Genetics and Evolution, University of Geneva, Sciences 3, 30, Quai Ernest Ansermet, CH-1211 Geneva 4, Switzerland IRCAM, UMR 9912, Université Pierre et Marie Curie, Paris, France
Department of Genetics and Evolution, University of Geneva, Sciences 3, 30, Quai Ernest Ansermet, CH-1211 Geneva 4, Switzerland.
Nucleic Acids Res. 2015 Mar 11;43(5):2513-24. doi: 10.1093/nar/gkv107. Epub 2015 Feb 17.
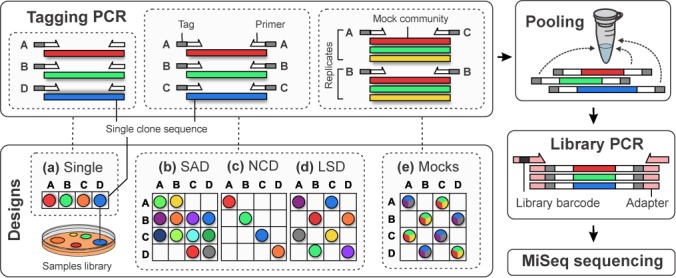
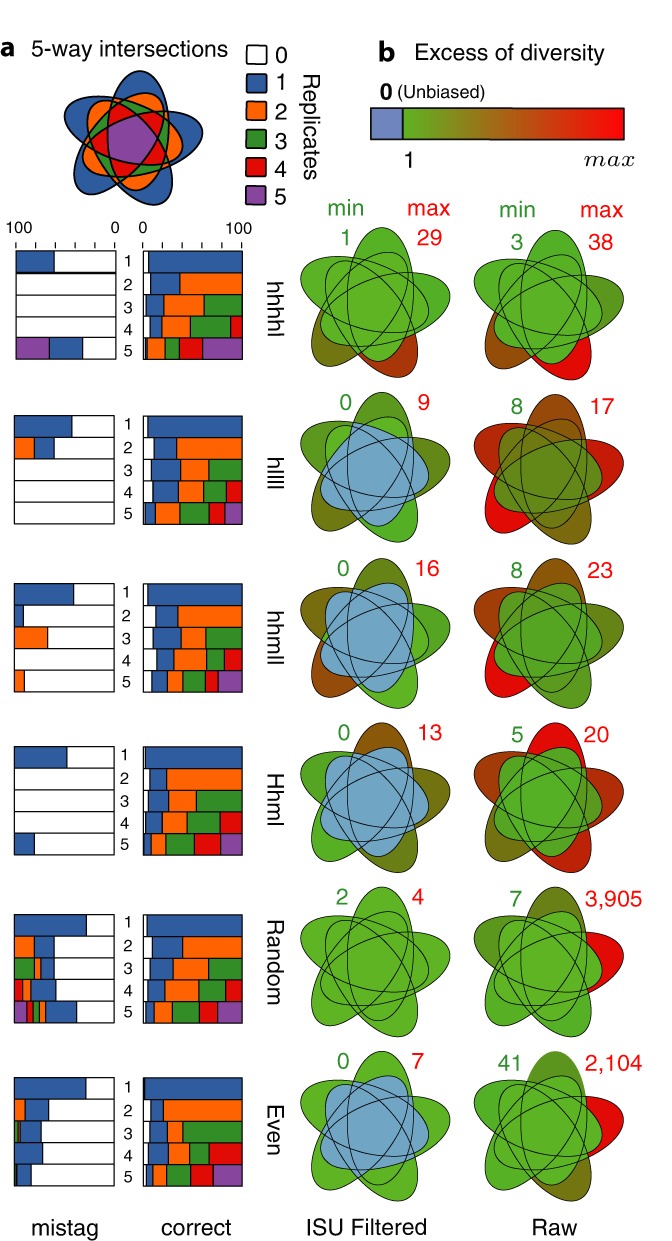
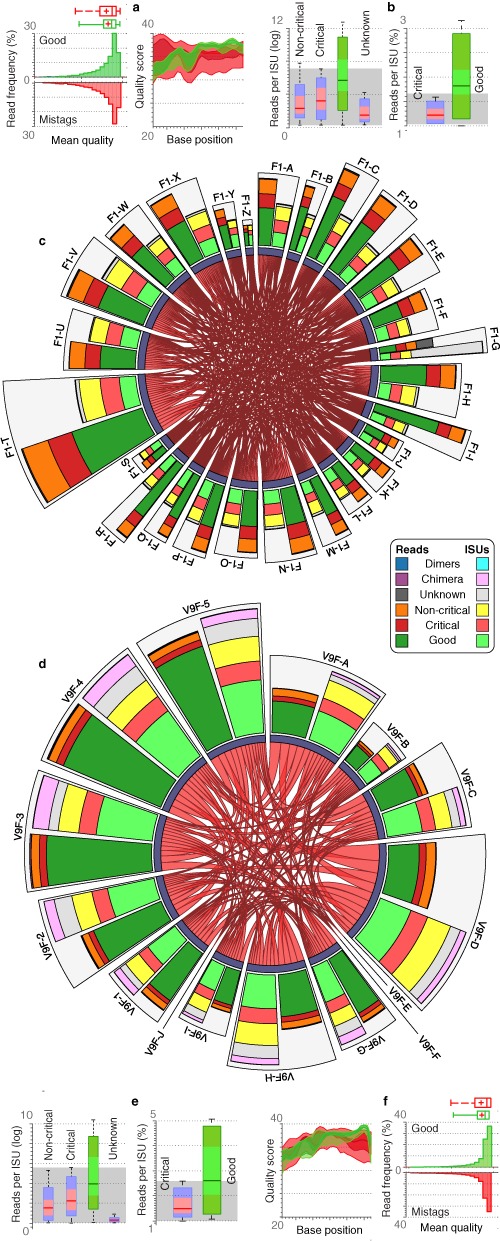
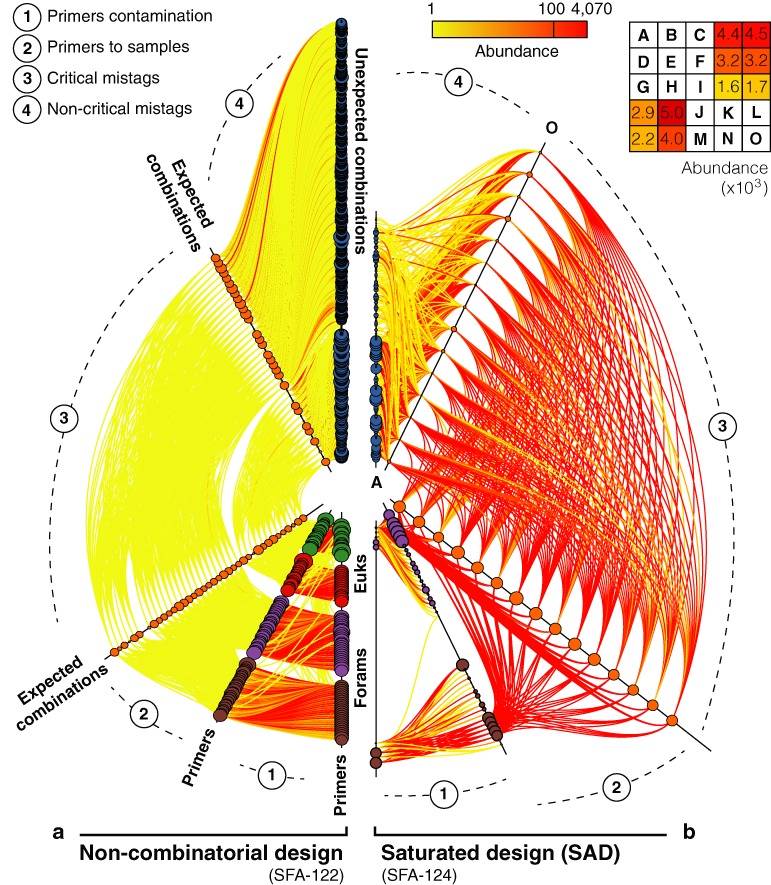
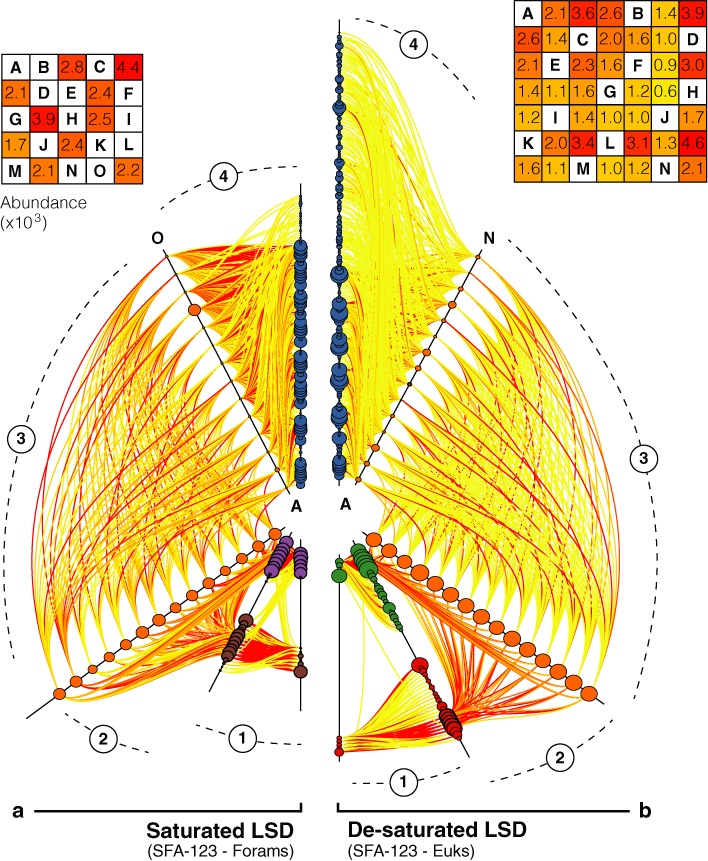
Tagging amplicons with tag sequences appended to PCR primers allow the multiplexing of numerous samples for high-throughput sequencing (HTS). This approach is routinely used in HTS-based diversity analyses, especially in microbial ecology and biomedical diagnostics. However, amplicon library preparation is subject to pervasive sample sequence cross-contaminations as a result of tag switching events referred to as mistagging. Here, we sequenced seven amplicon libraries prepared using various multiplexing designs in order to measure the magnitude of this phenomenon and its impact on diversity analyses. Up to 28.2% of the unique sequences correspond to undetectable (critical) mistags in single- or saturated double-tagging libraries. We show the advantage of multiplexing samples following Latin Square Designs in order to optimize the detection of mistags and maximize the information on their distribution across samples. We use this information in designs incorporating PCR replicates to filter the critical mistags and to recover the exact composition of mock community samples. Being parameter-free and data-driven, our approach can provide more accurate and reproducible HTS data sets, improving the reliability of their interpretations.
通过在PCR引物上附加标签序列来标记扩增子,能够对众多样本进行多重分析,以实现高通量测序(HTS)。这种方法在基于HTS的多样性分析中经常使用,特别是在微生物生态学和生物医学诊断领域。然而,由于被称为错误标记的标签切换事件,扩增子文库制备容易受到普遍的样本序列交叉污染。在这里,我们对使用各种多重设计制备的七个扩增子文库进行了测序,以测量这种现象的严重程度及其对多样性分析的影响。在单标签或饱和双标签文库中,高达28.2%的独特序列对应于不可检测的(关键)错误标记。我们展示了按照拉丁方设计对样本进行多重分析的优势,以便优化错误标记的检测,并最大限度地获取其在样本间分布的信息。我们在包含PCR重复的设计中使用这些信息来过滤关键错误标记,并恢复模拟群落样本的准确组成。我们的方法无需参数且由数据驱动,可以提供更准确和可重复的HTS数据集,提高其解释的可靠性。