de Groot J R
Neth Heart J. 2002 Sep;10(9):360-365.
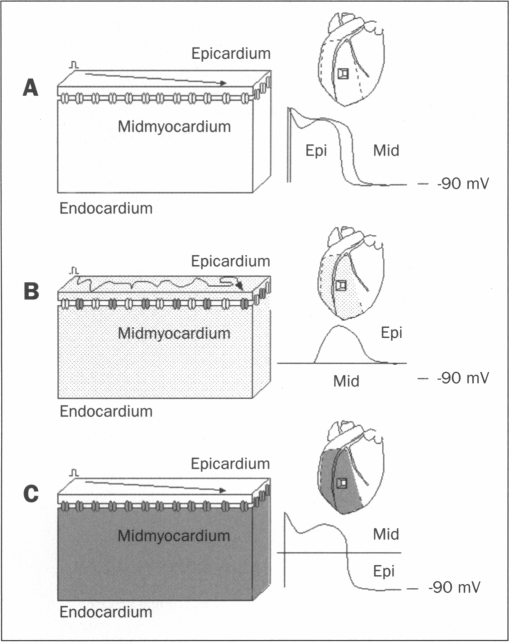
Sudden death resulting from ventricular fibrillation (VF) during acute myocardial ischaemia forms an important contribution to mortality associated with infarction. Its temporal distribution is not known, but 30% of mortality occurs within the first 60 minutes. Two distinct phases of arrhythmias have been demonstrated in laboratory animals subjected to coronary occlusion. The mechanism of the second, 1B phase (which is associated with more lethal events than the first, 1A phase) is largely unknown but appears to be related to cellular uncoupling, i.e. the closure of gap junctions. Gap junctions are intercellular communication channels that are permeable for ions and metabolites and are necessary for normal propagation of electrical activation. It has been suggested that closure of gap junctions results in a largely inhomogeneous substrate in which microreentry forms the electrophysiological mechanism for VF. However, there is growing support for the hypothesis that arrhythmias relate to the persistence of residual coupling rather than to the occurrence of uncoupling. With this, the ischaemic midmyocardium can depress the intrinsically viable tissue of the ischaemic subepicardium and subendocardium and cause conduction slowing and block leading to arrhythmias. Progression of uncoupling terminates this interaction and allows the subepicardium and subendocardium to recover. Indeed, electrophysiological properties recover subepicardially whereas the midmyocardial tissue becomes inexcitable. In addition, activation patterns during VF become restricted to the two-dimensional plane of the subepicardium. These observations support the hypothesis of residual coupling as an arrhythmogenic mechanism during the delayed phase of acute ischaemia. Whether this mechanism is equally important in patients with remodelled and failing hearts can at this time only be speculated upon. However, modifying intercellular coupling might turn out a new antiarrhythmic therapy.
急性心肌缺血期间室颤(VF)导致的猝死是梗死相关死亡率的重要组成部分。其时间分布尚不清楚,但30%的死亡发生在最初60分钟内。在冠状动脉闭塞的实验动物中已证实存在两个不同的心律失常阶段。第二个阶段即1B期(与比第一个阶段即1A期更致命的事件相关)的机制很大程度上未知,但似乎与细胞解偶联有关,即缝隙连接的关闭。缝隙连接是细胞间通讯通道,对离子和代谢产物具有通透性,是电活动正常传播所必需的。有人提出,缝隙连接的关闭会导致一个很大程度上不均匀的基质,其中微折返形成室颤的电生理机制。然而,越来越多的人支持这样一种假说,即心律失常与残余耦联的持续存在有关,而不是与解偶联的发生有关。据此,缺血心肌中层可抑制缺血心外膜下层和心内膜下层的内在存活组织,并导致传导减慢和阻滞,从而引发心律失常。解偶联的进展终止这种相互作用,并使心外膜下层和心内膜下层得以恢复。事实上,心外膜下层的电生理特性恢复,而心肌中层组织变得不可兴奋。此外,室颤期间的激活模式局限于心外膜下层的二维平面。这些观察结果支持残余耦联作为急性缺血延迟期致心律失常机制的假说。目前只能推测这种机制在心脏重塑和衰竭患者中是否同样重要。然而,改变细胞间耦联可能会成为一种新的抗心律失常治疗方法。