Woo J A, Zhao X, Khan H, Penn C, Wang X, Joly-Amado A, Weeber E, Morgan D, Kang D E
1] Department of Molecular Medicine, University of South Florida Morsani College of Medicine, Tampa, FL, USA [2] USF Health Byrd Alzheimer's Institute, University of South Florida Morsani College of Medicine, Tampa, FL, USA.
USF Health Byrd Alzheimer's Institute, University of South Florida Morsani College of Medicine, Tampa, FL, USA.
Cell Death Differ. 2015 Jun;22(6):921-34. doi: 10.1038/cdd.2015.5. Epub 2015 Feb 20.
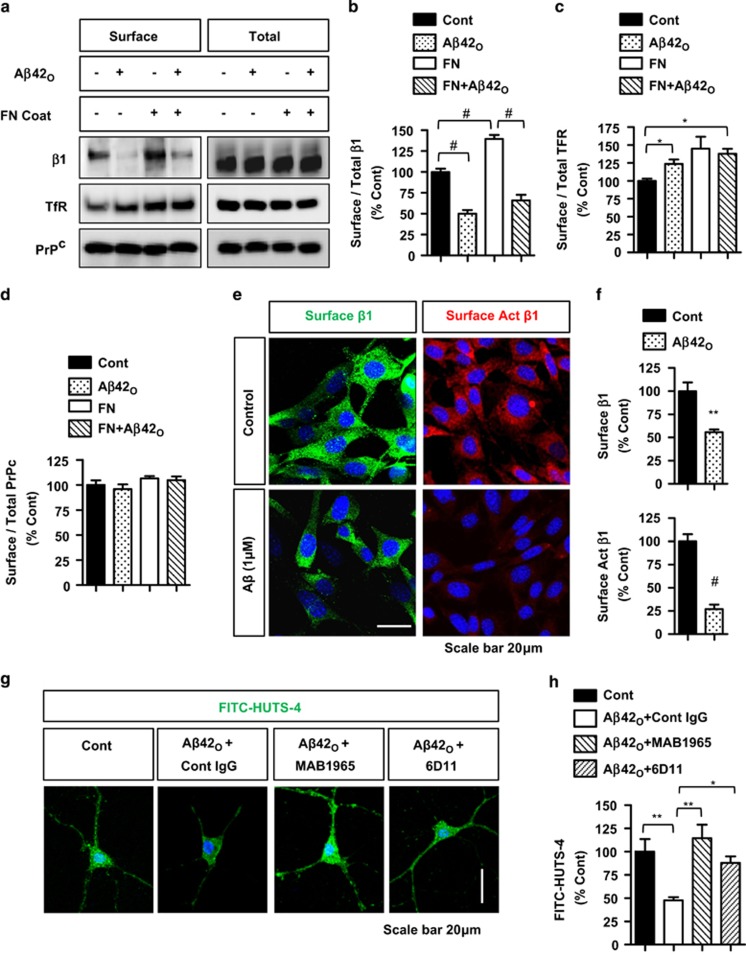
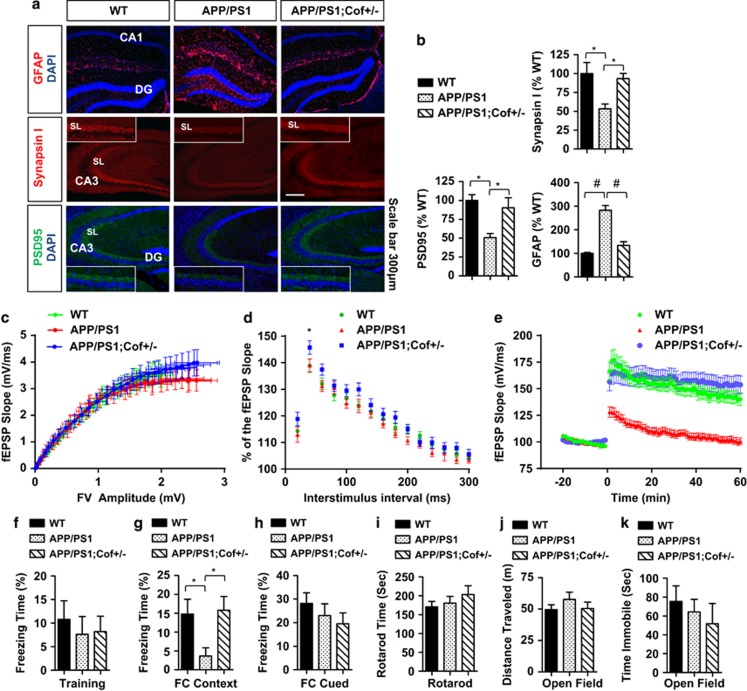
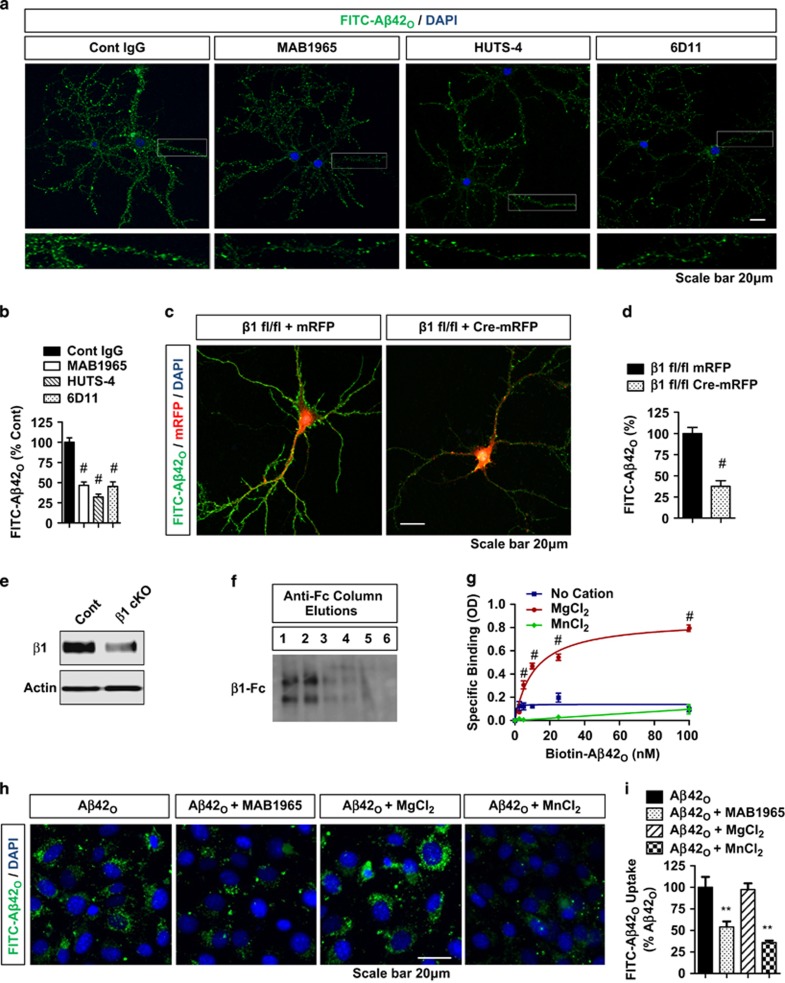
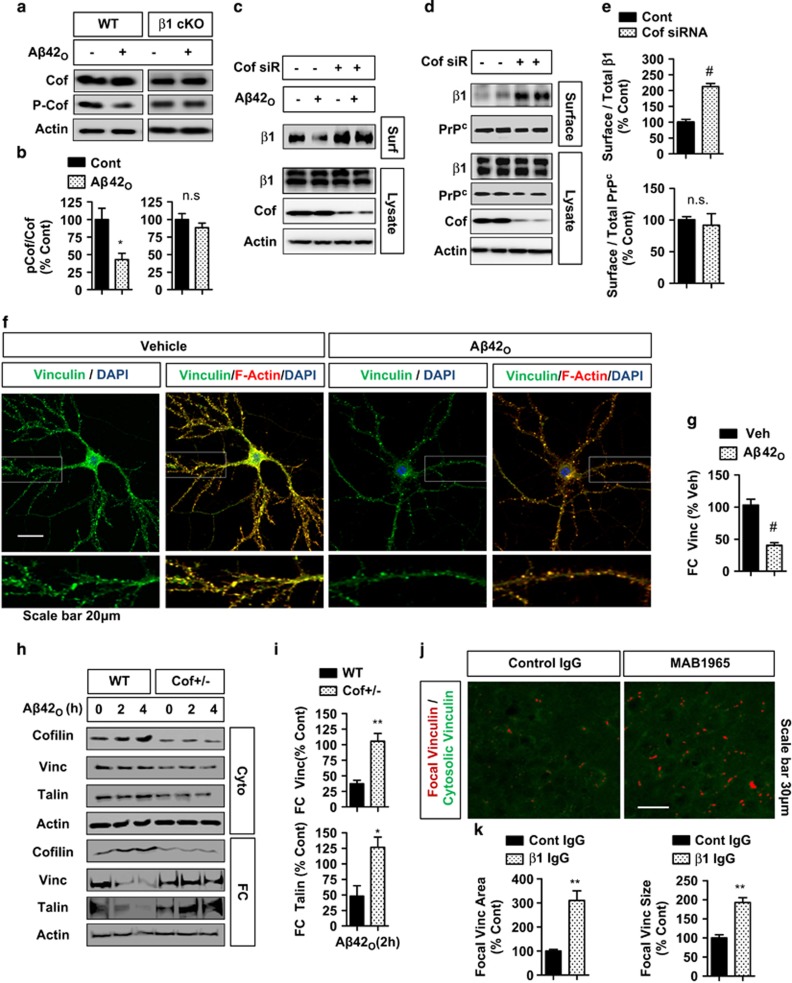
The accumulation of amyloid-β protein (Aβ) is an early event associated with synaptic and mitochondrial damage in Alzheimer's disease (AD). Recent studies have implicated the filamentous actin (F-actin) severing protein, Cofilin, in synaptic remodeling, mitochondrial dysfunction, and AD pathogenesis. However, whether Cofilin is an essential component of the AD pathogenic process and how Aβ impinges its signals to Cofilin from the neuronal surface are unknown. In this study, we found that Aβ42 oligomers (Aβ42O, amyloid-β protein 1-42 oligomers) bind with high affinity to low or intermediate activation conformers of β1-integrin, resulting in the loss of surface β1-integrin and activation of Cofilin via Slingshot homology-1 (SSH1) activation. Specifically, conditional loss of β1-integrin prevented Aβ42O-induced Cofilin activation, and allosteric modulation or activation of β1-integrin significantly reduced Aβ42O binding to neurons while blocking Aβ42O-induced reactive oxygen species (ROS) production, mitochondrial dysfunction, depletion of F-actin/focal Vinculin, and apoptosis. Cofilin, in turn, was required for Aβ42O-induced loss of cell surface β1-integrin, disruption of F-actin/focal Talin-Vinculin, and depletion of F-actin-associated postsynaptic proteins. SSH1 reduction, which mitigated Cofilin activation, prevented Aβ42O-induced mitochondrial Cofilin translocation and apoptosis, while AD brain mitochondria contained significantly increased activated/oxidized Cofilin. In mechanistic support in vivo, AD mouse model (APP (amyloid precursor protein)/PS1) brains contained increased SSH1/Cofilin and decreased SSH1/14-3-3 complexes, indicative of SSH1-Cofilin activation via release of SSH1 from 14-3-3. Finally, genetic reduction in Cofilin rescued APP/Aβ-induced synaptic protein loss and gliosis in vivo as well as deficits in long-term potentiation (LTP) and contextual memory in APP/PS1 mice. These novel findings therefore implicate the essential involvement of the β1-integrin-SSH1-Cofilin pathway in mitochondrial and synaptic dysfunction in AD.
淀粉样β蛋白(Aβ)的积累是阿尔茨海默病(AD)中与突触和线粒体损伤相关的早期事件。最近的研究表明,丝状肌动蛋白(F-肌动蛋白)切断蛋白Cofilin与突触重塑、线粒体功能障碍及AD发病机制有关。然而,Cofilin是否为AD致病过程的关键组成部分,以及Aβ如何从神经元表面将其信号传递给Cofilin尚不清楚。在本研究中,我们发现Aβ42寡聚体(Aβ42O,淀粉样β蛋白1-42寡聚体)与β1整合素的低或中等激活构象具有高亲和力结合,导致表面β1整合素丧失,并通过弹弓同源物1(SSH1)激活来激活Cofilin。具体而言,β1整合素的条件性缺失可阻止Aβ42O诱导的Cofilin激活,β1整合素的变构调节或激活可显著减少Aβ42O与神经元的结合,同时阻断Aβ42O诱导的活性氧(ROS)产生、线粒体功能障碍、F-肌动蛋白/粘着斑纽蛋白耗竭及细胞凋亡。反过来,Cofilin是Aβ42O诱导细胞表面β1整合素丧失、F-肌动蛋白/粘着斑塔林-纽蛋白破坏及F-肌动蛋白相关突触后蛋白耗竭所必需的。SSH1减少可减轻Cofilin激活,从而防止Aβ42O诱导的线粒体Cofilin易位和细胞凋亡,而AD脑线粒体中活化/氧化型Cofilin显著增加。在体内机制支持方面,AD小鼠模型(APP(淀粉样前体蛋白)/PS1)脑内SSH1/Cofilin增加,SSH1/14-3-3复合物减少,表明SSH1通过从14-3-3释放而激活Cofilin。最后,Cofilin的基因敲低可挽救APP/Aβ诱导的体内突触蛋白丢失和胶质增生,以及APP/PS1小鼠的长时程增强(LTP)和情境记忆缺陷。因此,这些新发现表明β1整合素-SSH1-Cofilin途径在AD的线粒体和突触功能障碍中起重要作用。