Division of Infectious Diseases , Brigham and Women's Hospital , Boston, Massachusetts ; Harvard Medical School , Boston, Massachusetts.
École Polytechnique Fédérale de Lausanne and University of Lausanne , Switzerland ; University Hospital and University of Lausanne , Switzerland ; Swiss Institute of Bioinformatics , Switzerland.
Open Forum Infect Dis. 2014 May 22;1(1):ofu018. doi: 10.1093/ofid/ofu018. eCollection 2014 Mar.
We conducted a genome-wide association study to explore whether common host genetic variants (>5% frequency) were associated with presence of virus able to use CXCR4 for entry.
Phenotypic determination of human immunodeficiency virus (HIV)-1 coreceptor usage was performed on pretreatment plasma HIV-1 samples from treatment-naive participants in AIDS Clinical Trials Group A5095, a study of initial antiretroviral regimens. Associations between genome-wide single-nucleotide polymorphisms (SNPs), CCR5 Δ32 genotype, and human leukocyte antigen (HLA) class I alleles and viral coreceptor usage were explored.
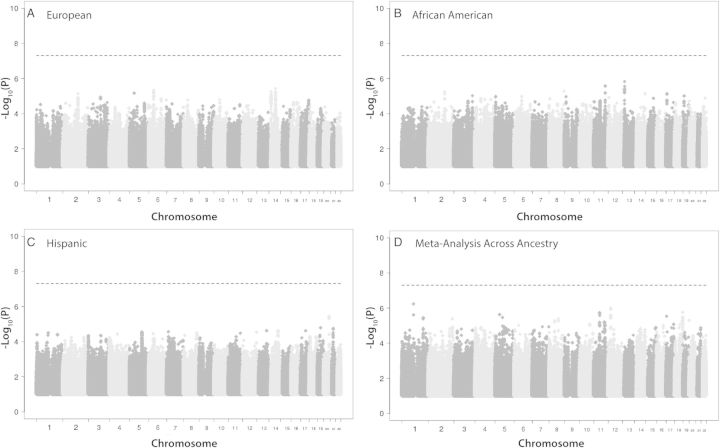
Viral phenotypes were obtained from 593 patients with available genome-wide SNP data. Forty-four percent of subjects had virus capable of using CXCR4 for entry as determined by phenotyping. Overall, no associations, including those between polymorphisms in genes encoding viral coreceptors and their promoter regions or in HLA genes previously associated with HIV-1 disease progression, passed the statistical threshold for genome-wide significance (P < 5.0 × 10(-8)) in any comparison. However, the presence of viruses able to use CXCR4 for entry was marginally associated with the CCR5 Δ32 genotype in the nongenome-wide analysis.
No human genetic variants were significantly associated with virus able to use CXCR4 for entry at the genome-wide level. Although the sample size had limited power to definitively exclude genetic associations, these results suggest that host genetic factors, including those that influence coreceptor expression or the immune pressures leading to viral envelope diversity, are either rare or have only modest effects in determining HIV-1 coreceptor usage.
我们进行了全基因组关联研究,以探索常见的宿主遗传变异(频率>5%)是否与能够利用 CXCR4 进入的病毒的存在相关。
在 AIDS Clinical Trials Group A5095 中对未经治疗的参与者的预处理血浆 HIV-1 样本进行了人类免疫缺陷病毒(HIV-1)辅助受体使用情况的表型测定,这是一项初始抗逆转录病毒方案的研究。探索了全基因组单核苷酸多态性(SNP)、CCR5 Δ32 基因型和人类白细胞抗原(HLA)I 类等位基因与病毒辅助受体使用之间的关联。
从 593 名具有可用全基因组 SNP 数据的患者中获得了病毒表型。44%的受试者的病毒能够通过表型测定利用 CXCR4 进入。总体而言,包括编码病毒辅助受体及其启动子区域的基因中的多态性以及先前与 HIV-1 疾病进展相关的 HLA 基因中的多态性在内的任何比较都没有通过全基因组显著水平(P < 5.0×10(-8))的统计阈值。然而,在非全基因组分析中,能够利用 CXCR4 进入的病毒的存在与 CCR5 Δ32 基因型呈边缘相关。
没有人类遗传变异与能够利用 CXCR4 进入的病毒在全基因组水平上显著相关。尽管样本量限制了确定遗传关联的能力,但这些结果表明,宿主遗传因素,包括影响辅助受体表达或导致病毒包膜多样性的免疫压力的因素,要么很少见,要么在确定 HIV-1 辅助受体使用方面仅有适度的影响。