Vlantis Katerina, Polykratis Apostolos, Welz Patrick-Simon, van Loo Geert, Pasparakis Manolis, Wullaert Andy
Institute for Genetics, University of Cologne, Cologne, Germany Centre for Molecular Medicine (CMMC), University of Cologne, Cologne, Germany Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD), University of Cologne, Cologne, Germany.
Institute for Genetics, University of Cologne, Cologne, Germany Centre for Molecular Medicine (CMMC), University of Cologne, Cologne, Germany Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD), University of Cologne, Cologne, Germany Institute for Research in Biomedicine (IRB), Barcelona, Spain.
Gut. 2016 Jun;65(6):935-43. doi: 10.1136/gutjnl-2014-308323. Epub 2015 Mar 11.
The gut microbiota modulates host susceptibility to intestinal inflammation, but the cell types and the signalling pathways orchestrating this bacterial regulation of intestinal homeostasis remain poorly understood. Here, we investigated the function of intestinal epithelial toll-like receptor (TLR) responses in the dextran sodium sulfate (DSS)-induced mouse model of colitis.
We applied an in vivo genetic approach allowing intestinal epithelial cell (IEC)-specific deletion of the critical TLR signalling adaptors, MyD88 and/or TIR-domain-containing adapter-inducing interferon-β (TRIF), as well as the downstream ubiquitin ligase TRAF6 in order to reveal the IEC-intrinsic function of these TLR signalling molecules during DSS colitis.
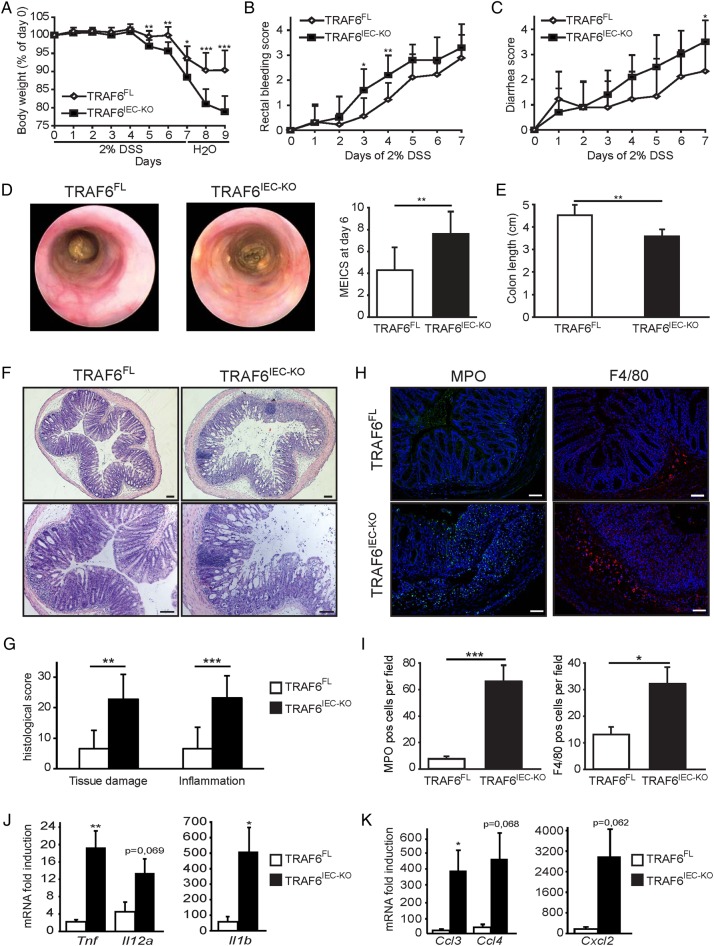
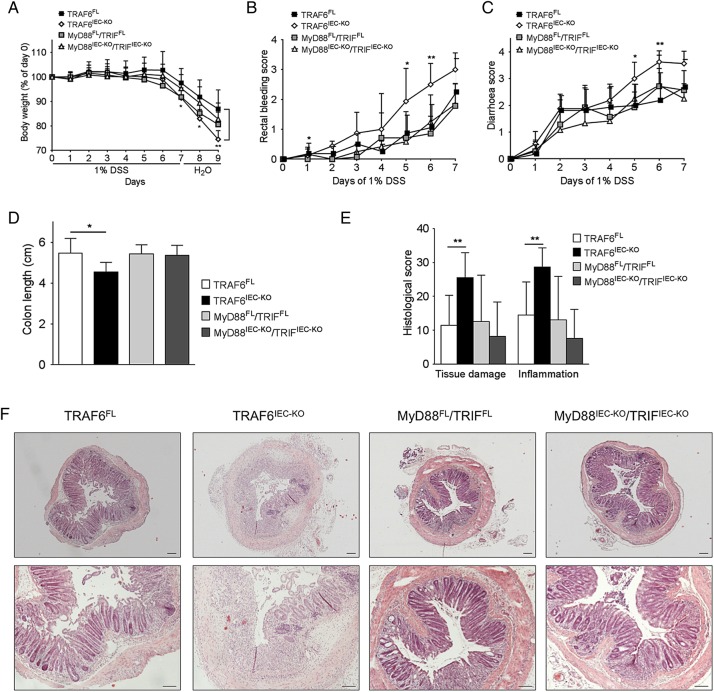
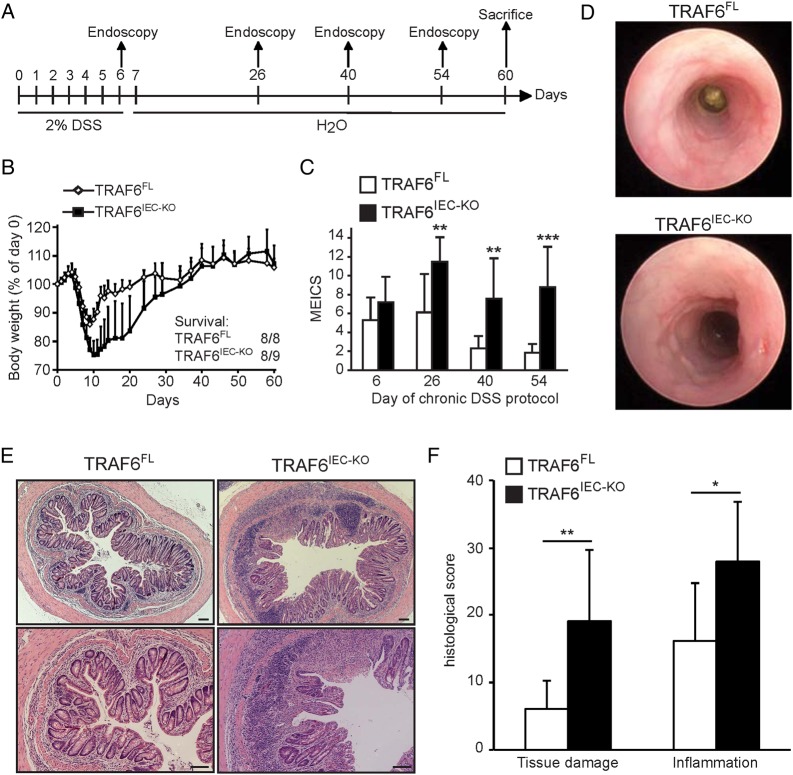
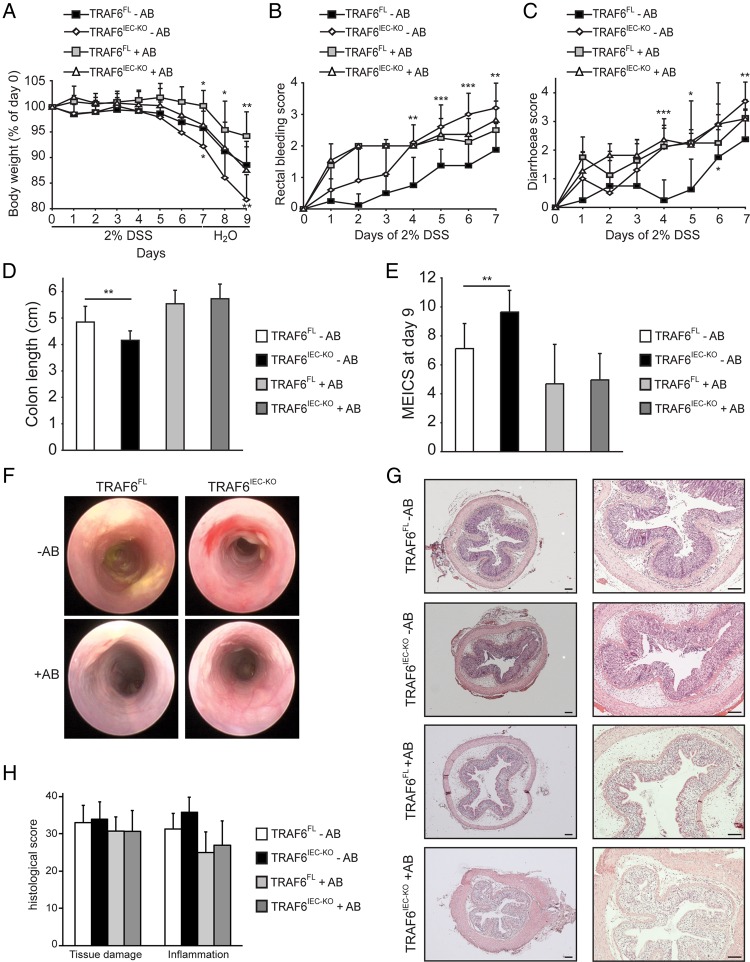
Mice lacking TRAF6 in IECs showed exacerbated DSS-induced inflammatory responses that ensued in the development of chronic colon inflammation. Antibiotic pretreatment abolished the increased DSS susceptibility of these mice, showing that epithelial TRAF6 signalling pathways prevent the gut microbiota from driving excessive colitis. However, in contrast to epithelial TRAF6 deletion, blocking epithelial TLR signalling by simultaneous deletion of MyD88 and TRIF specifically in IECs did not affect DSS-induced colitis severity. This in vivo functional comparison between TRAF6 and MyD88/TRIF deletion in IECs shows that the colitis-protecting effects of epithelial TRAF6 signalling are not triggered by TLRs.
Intestinal epithelial TRAF6-dependent but MyD88/TRIF-independent and, thus, TLR-independent signalling pathways are critical for preventing propagation of DSS-induced colon inflammation by the gut microbiota. Moreover, our experiments using mice with dual MyD88/TRIF deletion in IECs unequivocally show that the gut microbiota trigger non-epithelial TLRs rather than epithelial TLRs to restrict DSS colitis severity.
肠道微生物群调节宿主对肠道炎症的易感性,但协调这种细菌对肠道稳态调节的细胞类型和信号通路仍知之甚少。在此,我们在葡聚糖硫酸钠(DSS)诱导的小鼠结肠炎模型中研究了肠道上皮Toll样受体(TLR)反应的功能。
我们采用体内遗传学方法,特异性敲除肠道上皮细胞(IEC)中关键的TLR信号衔接蛋白MyD88和/或含TIR结构域的衔接蛋白诱导干扰素-β(TRIF),以及下游泛素连接酶TRAF6,以揭示这些TLR信号分子在DSS结肠炎期间的IEC内在功能。
IEC中缺乏TRAF6的小鼠表现出DSS诱导的炎症反应加剧,进而发展为慢性结肠炎症。抗生素预处理消除了这些小鼠对DSS易感性的增加,表明上皮TRAF6信号通路可防止肠道微生物群引发过度的结肠炎。然而,与上皮TRAF6缺失相反,通过在IEC中同时特异性敲除MyD88和TRIF来阻断上皮TLR信号,并不影响DSS诱导的结肠炎严重程度。这种IEC中TRAF6与MyD88/TRIF缺失的体内功能比较表明,上皮TRAF6信号的结肠炎保护作用不是由TLR触发的。
肠道上皮依赖TRAF6但不依赖MyD88/TRIF,因此不依赖TLR的信号通路对于防止肠道微生物群传播DSS诱导的结肠炎症至关重要。此外,我们使用IEC中MyD88/TRIF双缺失小鼠进行的实验明确表明,肠道微生物群触发非上皮TLR而非上皮TLR来限制DSS结肠炎的严重程度。