Boersma Vera, Moatti Nathalie, Segura-Bayona Sandra, Peuscher Marieke H, van der Torre Jaco, Wevers Brigitte A, Orthwein Alexandre, Durocher Daniel, Jacobs Jacqueline J L
Division of Molecular Oncology, The Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX, Amsterdam, The Netherlands.
The Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, 600 University Avenue, Toronto, ON M5G 1X5, Canada.
Nature. 2015 May 28;521(7553):537-540. doi: 10.1038/nature14216. Epub 2015 Mar 23.
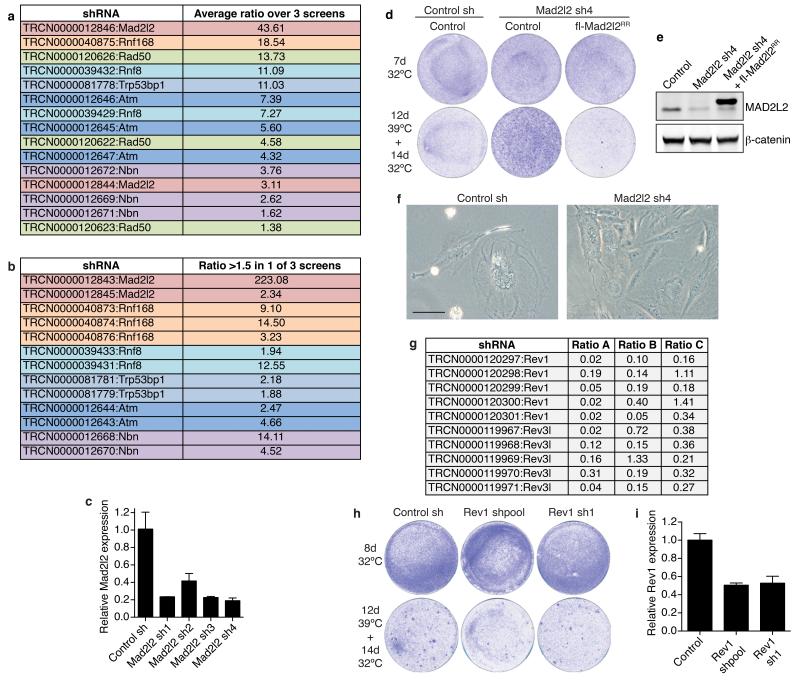
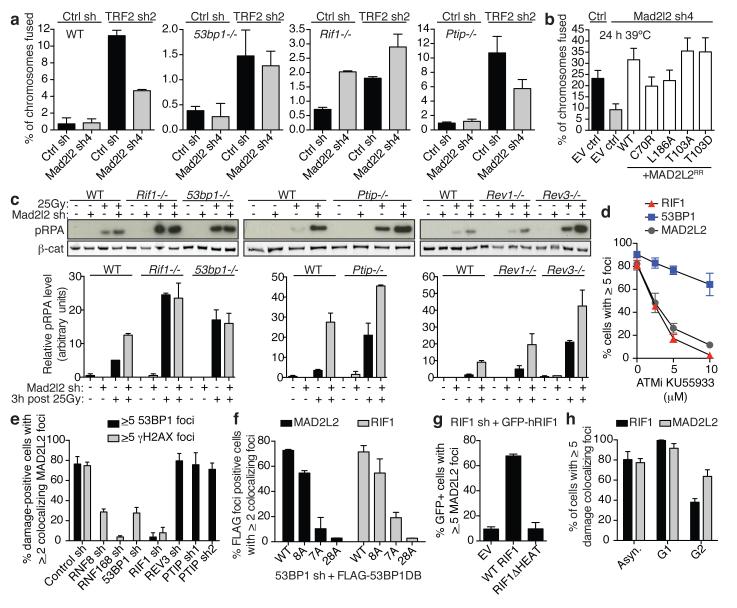
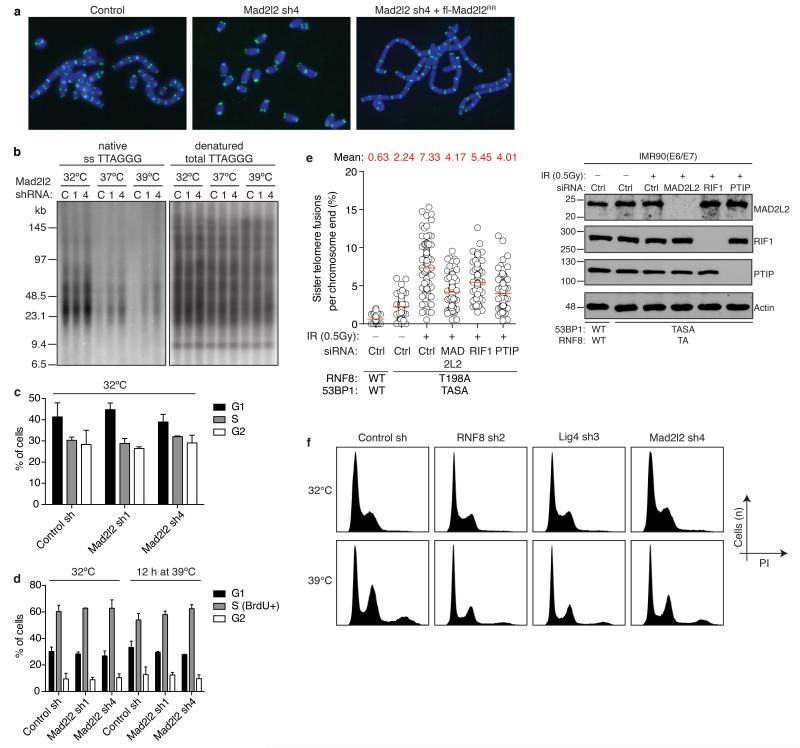
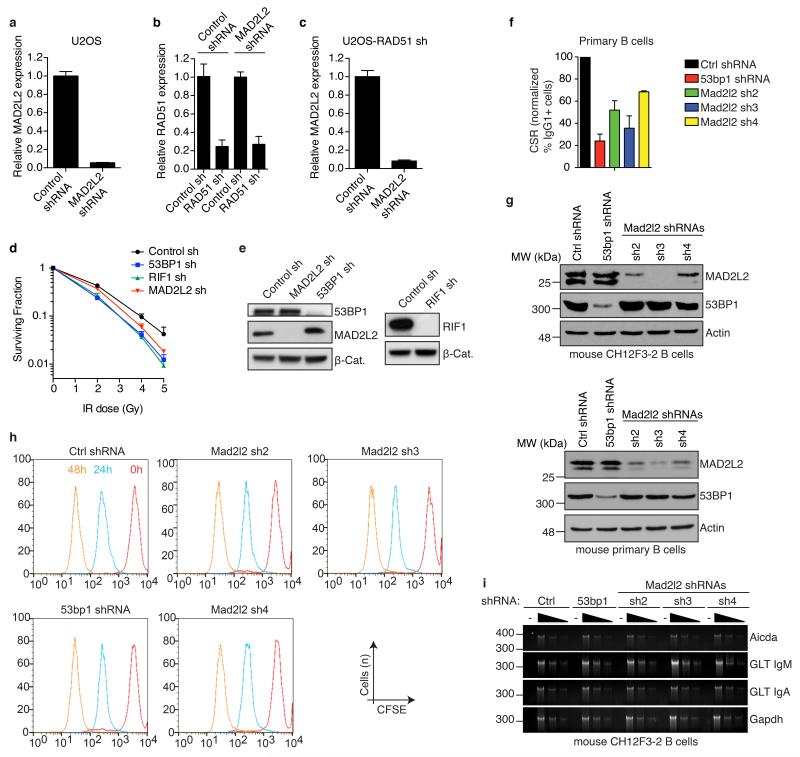
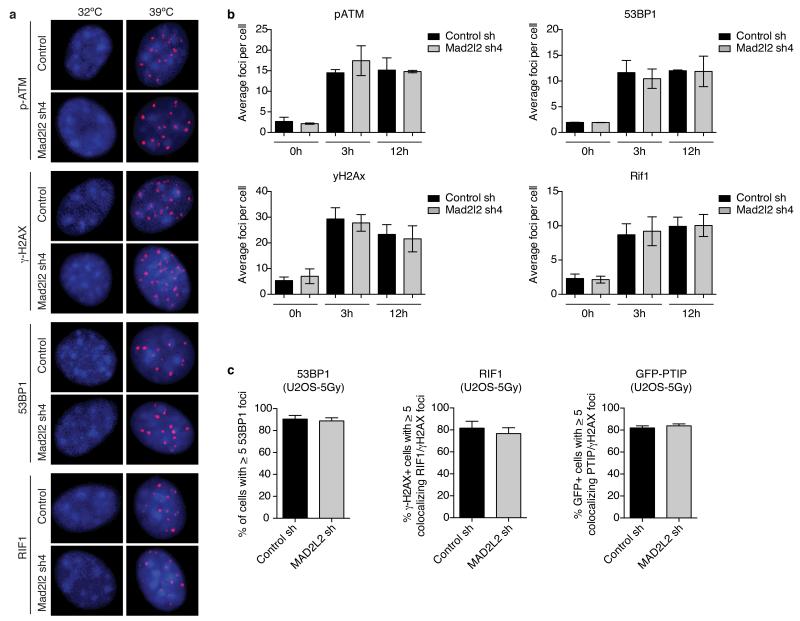
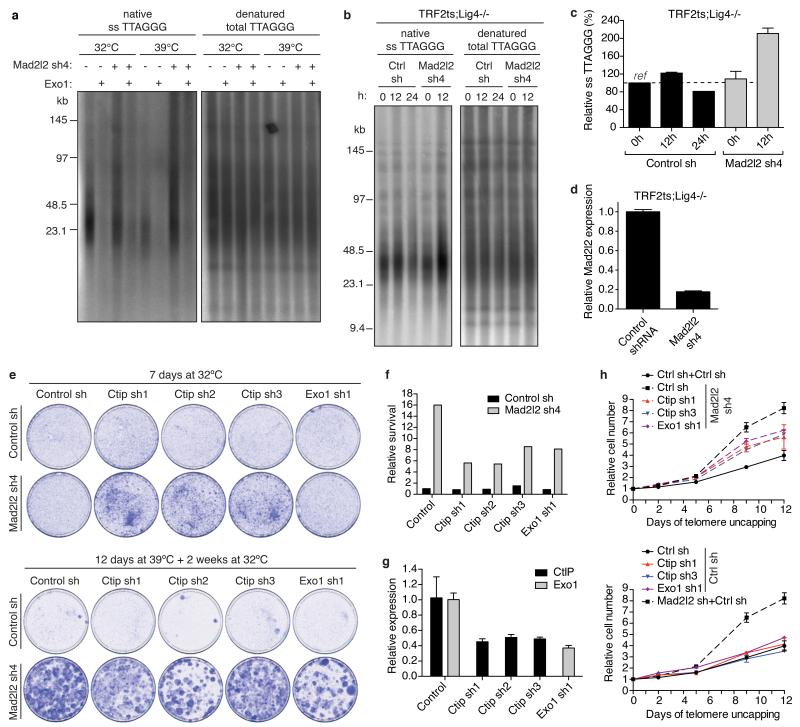
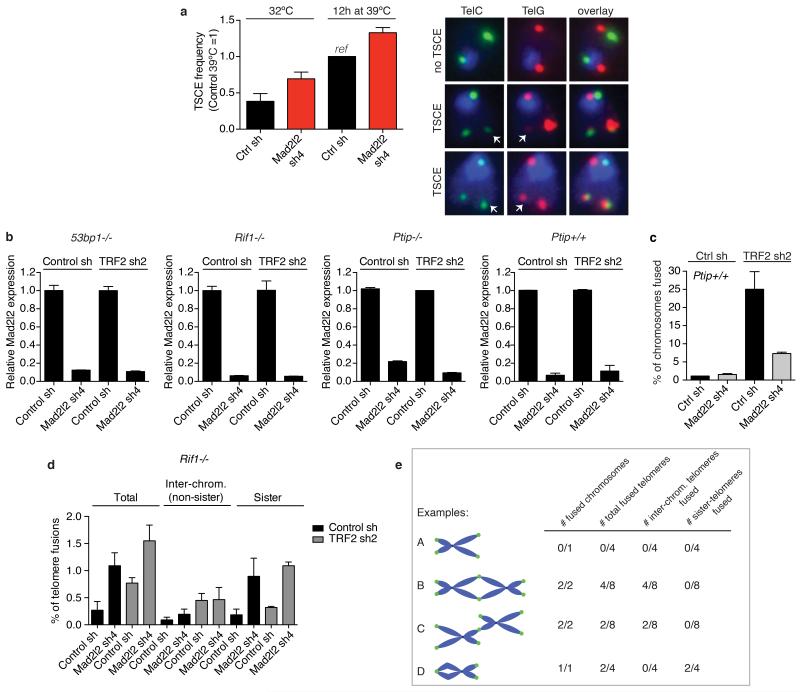
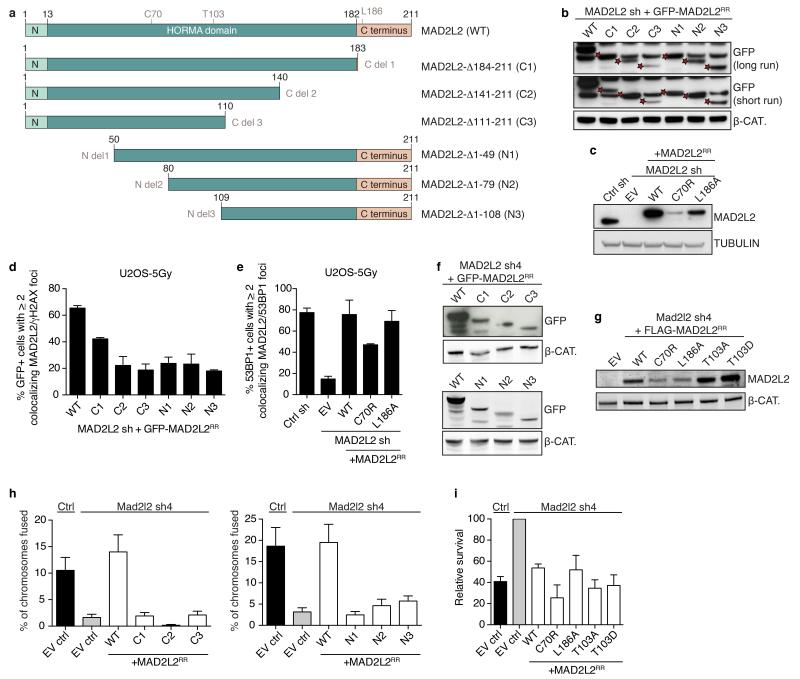
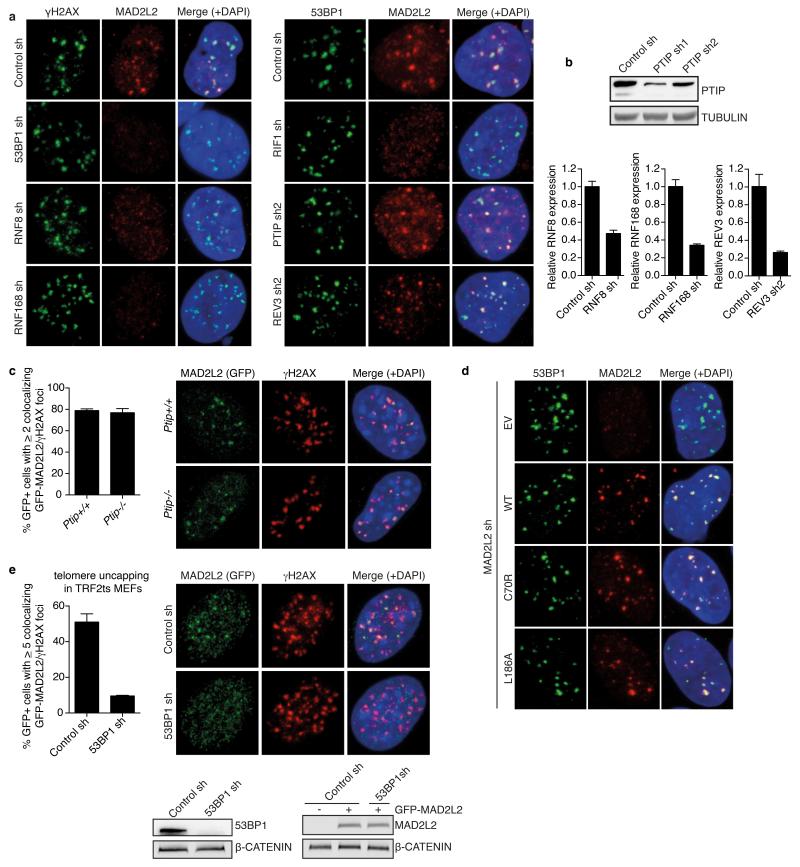
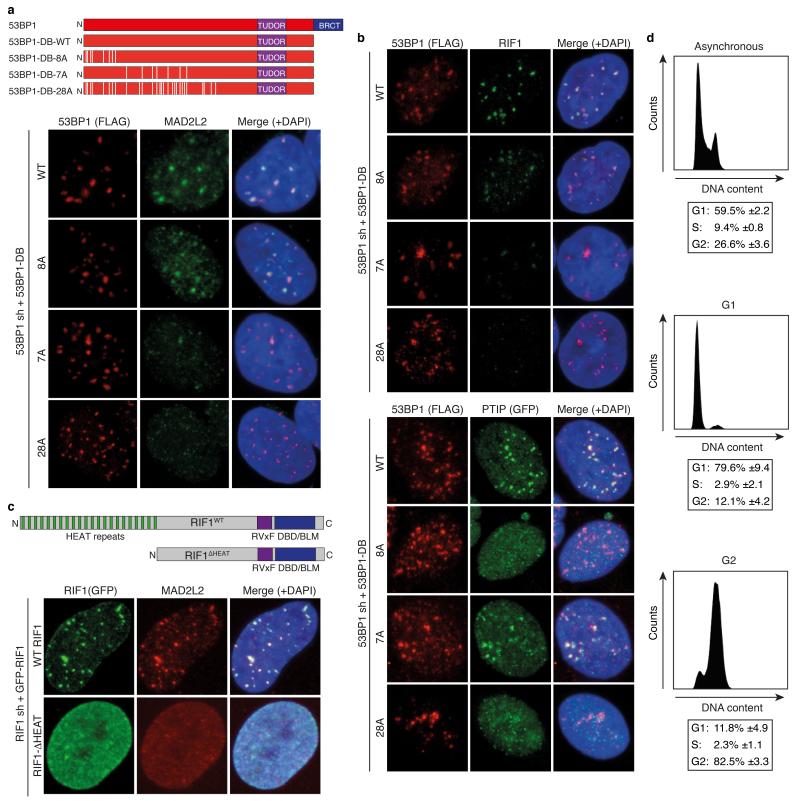
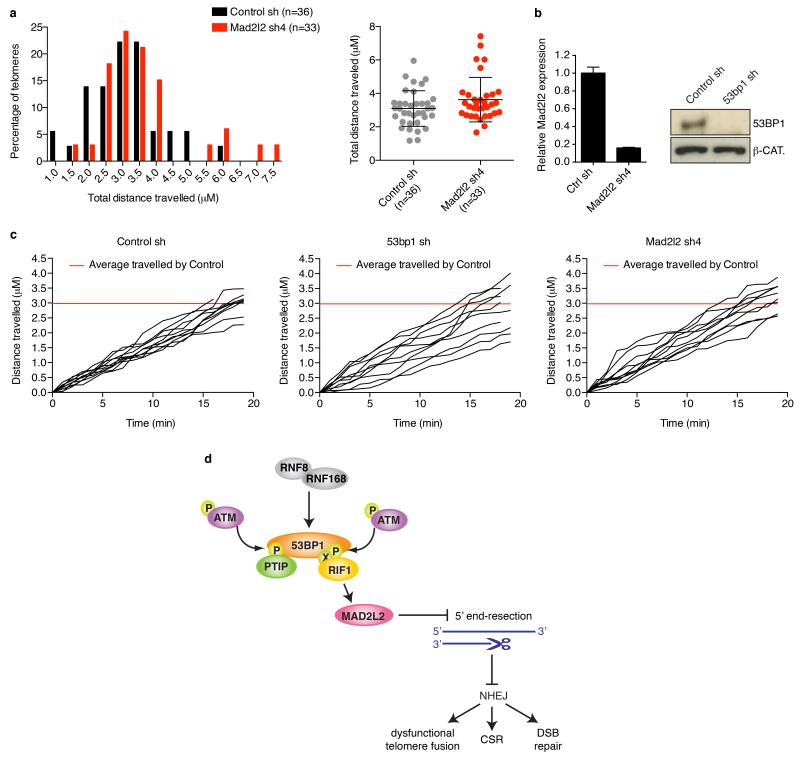
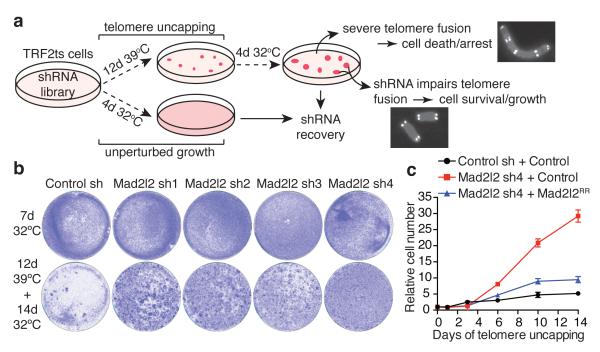
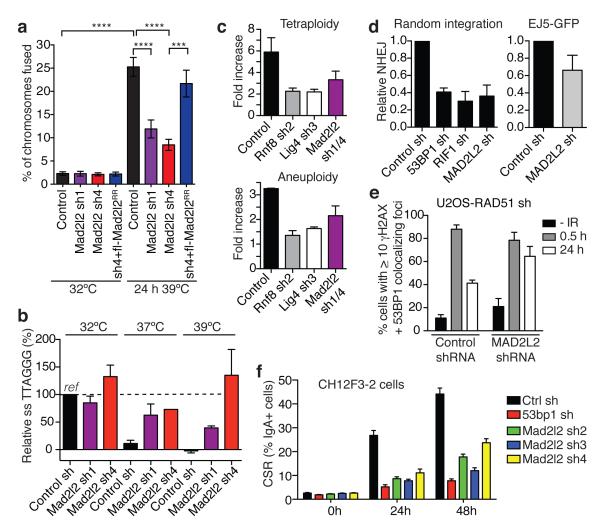
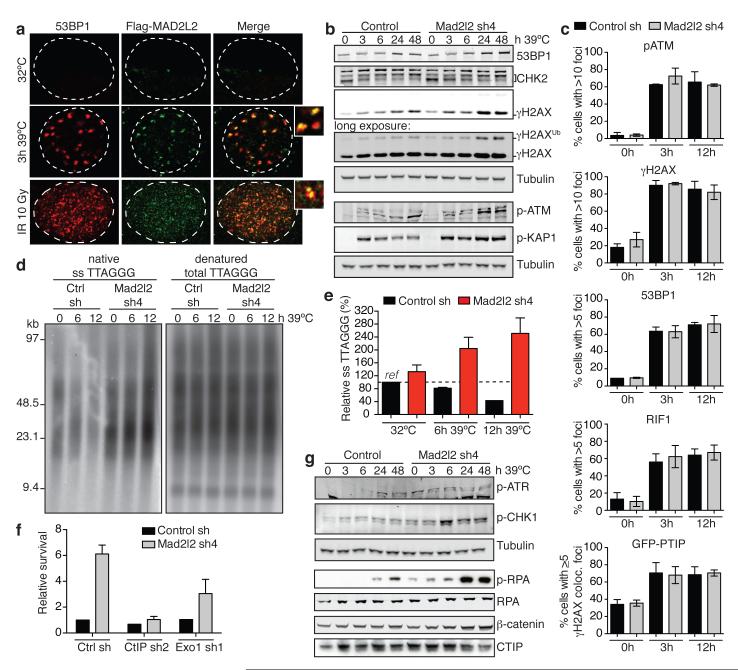
Appropriate repair of DNA lesions and the inhibition of DNA repair activities at telomeres are crucial to prevent genomic instability. By fuelling the generation of genetic alterations and by compromising cell viability, genomic instability is a driving force in cancer and ageing. Here we identify MAD2L2 (also known as MAD2B or REV7) through functional genetic screening as a novel factor controlling DNA repair activities at mammalian telomeres. We show that MAD2L2 accumulates at uncapped telomeres and promotes non-homologous end-joining (NHEJ)-mediated fusion of deprotected chromosome ends and genomic instability. MAD2L2 depletion causes elongated 3' telomeric overhangs, indicating that MAD2L2 inhibits 5' end resection. End resection blocks NHEJ while committing to homology-directed repair, and is under the control of 53BP1, RIF1 and PTIP. Consistent with MAD2L2 promoting NHEJ-mediated telomere fusion by inhibiting 5' end resection, knockdown of the nucleases CTIP or EXO1 partially restores telomere-driven genomic instability in MAD2L2-depleted cells. Control of DNA repair by MAD2L2 is not limited to telomeres. MAD2L2 also accumulates and inhibits end resection at irradiation-induced DNA double-strand breaks and promotes end-joining of DNA double-strand breaks in several settings, including during immunoglobulin class switch recombination. These activities of MAD2L2 depend on ATM kinase activity, RNF8, RNF168, 53BP1 and RIF1, but not on PTIP, REV1 and REV3, the latter two acting with MAD2L2 in translesion synthesis. Together, our data establish MAD2L2 as a crucial contributor to the control of DNA repair activity by 53BP1 that promotes NHEJ by inhibiting 5' end resection downstream of RIF1.
对DNA损伤进行适当修复以及抑制端粒处的DNA修复活性对于防止基因组不稳定至关重要。基因组不稳定通过促进基因改变的产生和损害细胞活力,成为癌症和衰老的驱动力。在这里,我们通过功能基因筛选鉴定出MAD2L2(也称为MAD2B或REV7)是控制哺乳动物端粒处DNA修复活性的一个新因子。我们表明,MAD2L2在无帽端粒处积累,并促进非同源末端连接(NHEJ)介导的去保护染色体末端融合和基因组不稳定。MAD2L2缺失导致3'端粒悬突延长,表明MAD2L2抑制5'端切除。末端切除会阻断NHEJ,同时致力于同源定向修复,并且受53BP1、RIF1和PTIP的控制。与MAD2L2通过抑制5'端切除促进NHEJ介导的端粒融合一致,核酸酶CTIP或EXO1的敲低部分恢复了MAD2L2缺失细胞中端粒驱动的基因组不稳定。MAD2L2对DNA修复的控制不仅限于端粒。MAD2L2在辐射诱导的DNA双链断裂处也会积累并抑制末端切除,并在多种情况下促进DNA双链断裂的末端连接,包括在免疫球蛋白类别转换重组期间。MAD2L2的这些活性依赖于ATM激酶活性、RNF8、RNF168、53BP1和RIF1,但不依赖于PTIP、REV1和REV3,后两者在跨损伤合成中与MAD2L2一起起作用。总之,我们的数据表明MAD2L2是53BP1控制DNA修复活性的关键因素,它通过抑制RIF1下游的5'端切除来促进NHEJ。