Harvey Michael G, Judy Caroline Duffie, Seeholzer Glenn F, Maley James M, Graves Gary R, Brumfield Robb T
Museum of Natural Science, Louisiana State University , Baton Rouge, LA , USA ; Department of Biological Sciences, Louisiana State University , Baton Rouge, LA , USA.
Museum of Natural Science, Louisiana State University , Baton Rouge, LA , USA ; Department of Biological Sciences, Louisiana State University , Baton Rouge, LA , USA ; Department of Vertebrate Zoology, MRC-116, National Museum of Natural History, Smithsonian Institution , Washington, D.C. , USA.
PeerJ. 2015 Apr 21;3:e895. doi: 10.7717/peerj.895. eCollection 2015.
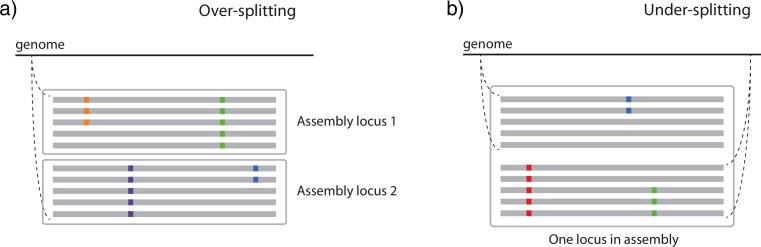
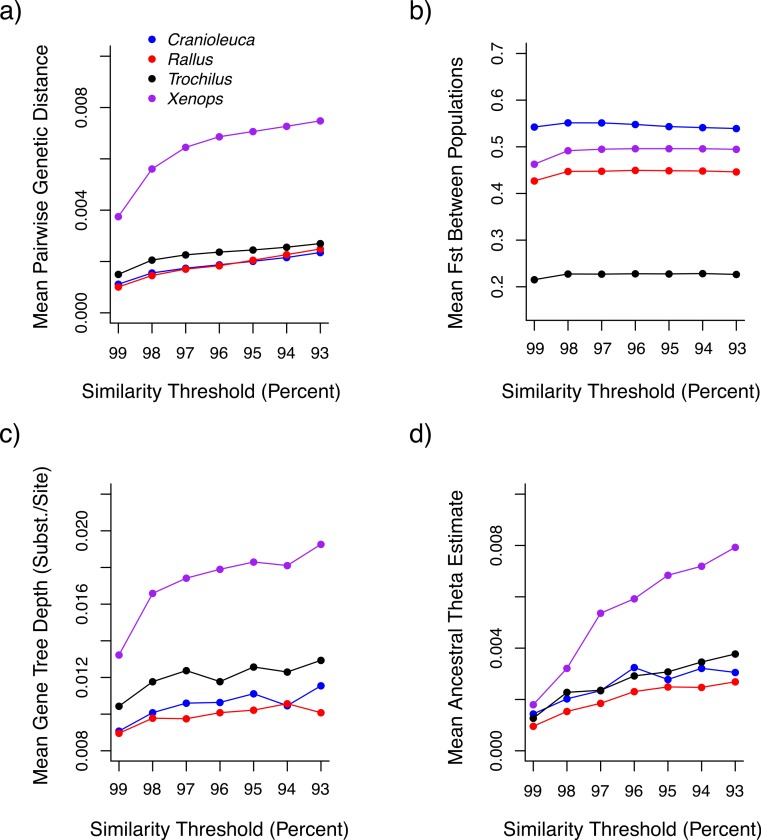
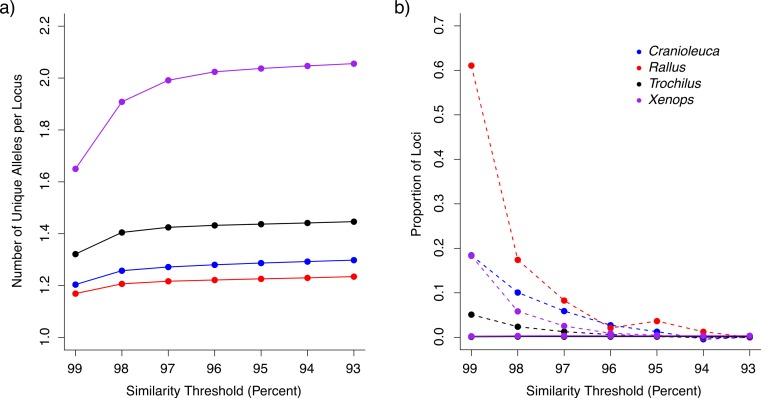
Comparing inferences among datasets generated using short read sequencing may provide insight into the concerted impacts of divergence, gene flow and selection across organisms, but comparisons are complicated by biases introduced during dataset assembly. Sequence similarity thresholds allow the de novo assembly of short reads into clusters of alleles representing different loci, but the resulting datasets are sensitive to both the similarity threshold used and to the variation naturally present in the organism under study. Thresholds that require high sequence similarity among reads for assembly (stringent thresholds) as well as highly variable species may result in datasets in which divergent alleles are lost or divided into separate loci ('over-splitting'), whereas liberal thresholds increase the risk of paralogous loci being combined into a single locus ('under-splitting'). Comparisons among datasets or species are therefore potentially biased if different similarity thresholds are applied or if the species differ in levels of within-lineage genetic variation. We examine the impact of a range of similarity thresholds on assembly of empirical short read datasets from populations of four different non-model bird lineages (species or species pairs) with different levels of genetic divergence. We find that, in all species, stringent similarity thresholds result in fewer alleles per locus than more liberal thresholds, which appears to be the result of high levels of over-splitting. The frequency of putative under-splitting, conversely, is low at all thresholds. Inferred genetic distances between individuals, gene tree depths, and estimates of the ancestral mutation-scaled effective population size (θ) differ depending upon the similarity threshold applied. Relative differences in inferences across species differ even when the same threshold is applied, but may be dramatically different when datasets assembled under different thresholds are compared. These differences not only complicate comparisons across species, but also preclude the application of standard mutation rates for parameter calibration. We suggest some best practices for assembling short read data to maximize comparability, such as using more liberal thresholds and examining the impact of different thresholds on each dataset.
比较使用短读长测序生成的数据集之间的推断,可能有助于深入了解分歧、基因流和选择对生物体的协同影响,但由于在数据集组装过程中引入的偏差,这种比较变得复杂。序列相似性阈值允许将短读长从头组装成代表不同位点的等位基因簇,但所得数据集对所使用的相似性阈值以及所研究生物体中自然存在的变异都很敏感。要求读长之间具有高序列相似性才能进行组装的阈值(严格阈值)以及高度可变的物种,可能会导致数据集中不同的等位基因丢失或被分成单独的位点(“过度拆分”),而宽松的阈值则增加了旁系同源位点被合并为单个位点的风险(“拆分不足”)。因此,如果应用不同的相似性阈值,或者物种在谱系内遗传变异水平上存在差异,那么数据集或物种之间的比较可能会存在偏差。我们研究了一系列相似性阈值对来自四个具有不同遗传分歧水平的不同非模式鸟类谱系(物种或物种对)群体的经验性短读长数据集组装的影响。我们发现,在所有物种中,严格的相似性阈值导致每个位点的等位基因数量比更宽松的阈值少,这似乎是过度拆分程度高的结果。相反,在所有阈值下,推测的拆分不足频率都很低。个体之间推断的遗传距离、基因树深度以及祖先突变尺度有效种群大小(θ)的估计值,取决于所应用的相似性阈值。即使应用相同的阈值,不同物种之间推断的相对差异也不同,但当比较在不同阈值下组装的数据集时,差异可能会非常显著。这些差异不仅使跨物种比较变得复杂,还排除了应用标准突变率进行参数校准的可能性。我们建议了一些组装短读长数据以最大化可比性的最佳做法,例如使用更宽松的阈值并检查不同阈值对每个数据集的影响。