Cocita Clément, Guiton Rachel, Bessou Gilles, Chasson Lionel, Boyron Marilyn, Crozat Karine, Dalod Marc
Centre d'Immunologie de Marseille-Luminy, UNIV UM2, Aix Marseille Université, Parc Scientifique et Technologique de Luminy, Marseille, France; Institut National de la Santé et de la Recherche Médicale (INSERM), U1104, Marseille, France; Centre National de la Recherche Scientifique (CNRS), UMR7280, Marseille, France.
PLoS Pathog. 2015 May 8;11(5):e1004897. doi: 10.1371/journal.ppat.1004897. eCollection 2015 May.
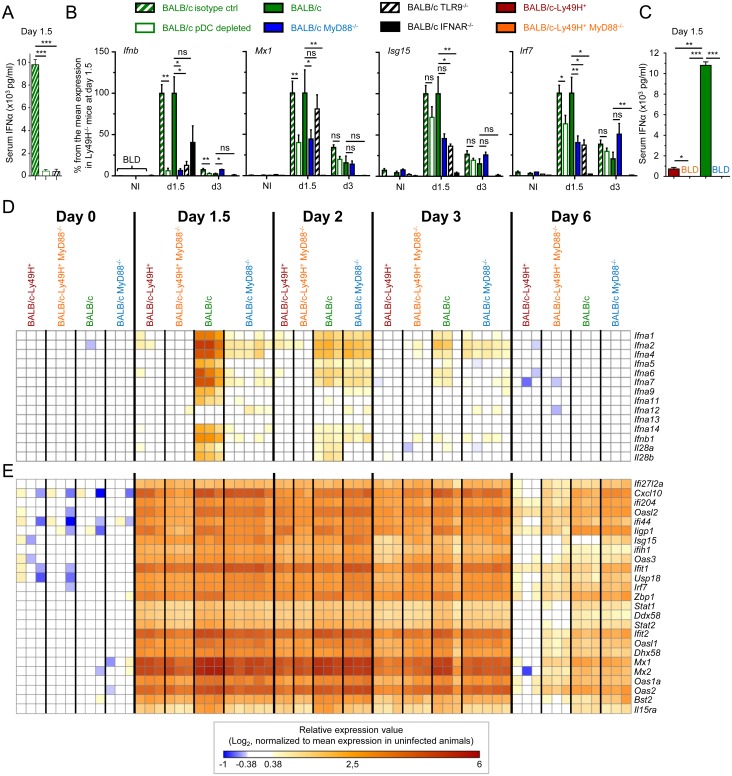
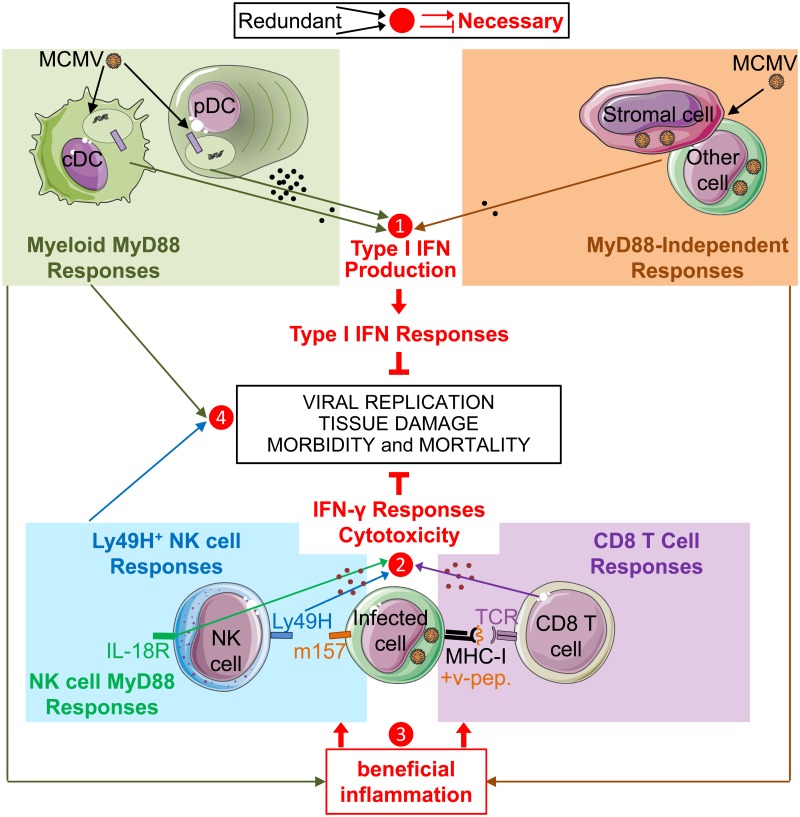
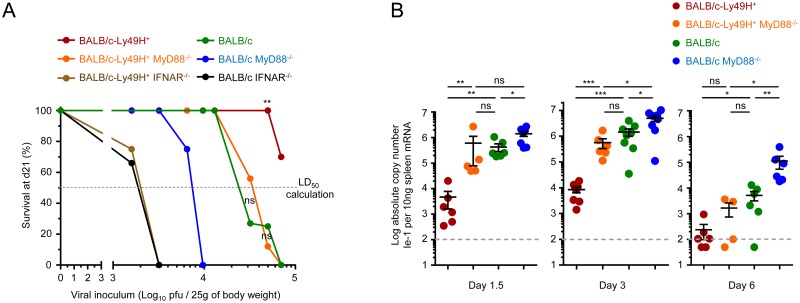
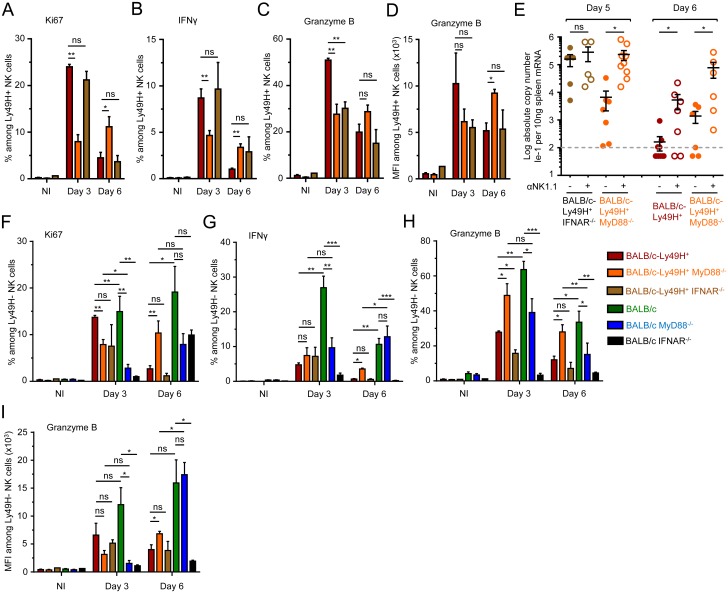
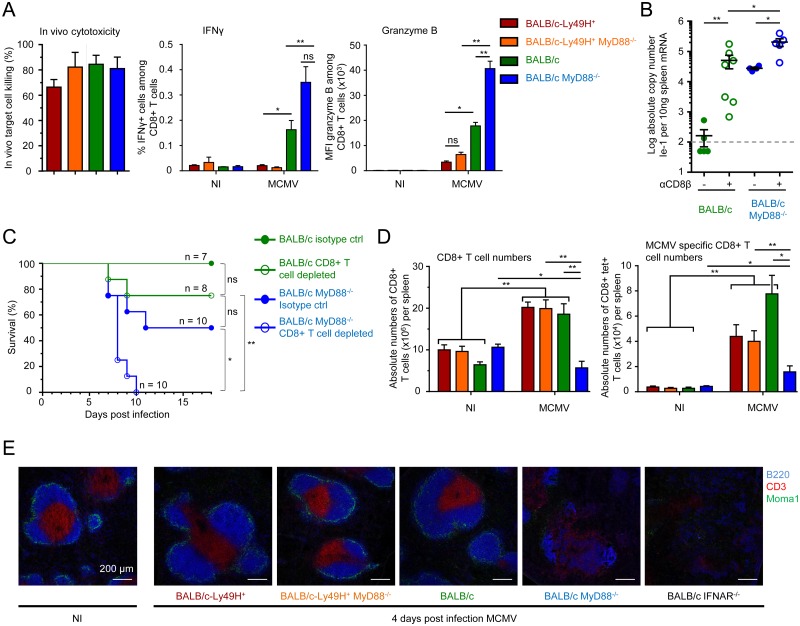
In mice, plasmacytoid dendritic cells (pDC) and natural killer (NK) cells both contribute to resistance to systemic infections with herpes viruses including mouse Cytomegalovirus (MCMV). pDCs are the major source of type I IFN (IFN-I) during MCMV infection. This response requires pDC-intrinsic MyD88-dependent signaling by Toll-Like Receptors 7 and 9. Provided that they express appropriate recognition receptors such as Ly49H, NK cells can directly sense and kill MCMV-infected cells. The loss of any one of these responses increases susceptibility to infection. However, the relative importance of these antiviral immune responses and how they are related remain unclear. In humans, while IFN-I responses are essential, MyD88 is dispensable for antiviral immunity. Hence, a higher redundancy has been proposed in the mechanisms promoting protective immune responses against systemic infections by herpes viruses during natural infections in humans. It has been assumed, but not proven, that mice fail to mount protective MyD88-independent IFN-I responses. In humans, the mechanism that compensates MyD88 deficiency has not been elucidated. To address these issues, we compared resistance to MCMV infection and immune responses between mouse strains deficient for MyD88, the IFN-I receptor and/or Ly49H. We show that selective depletion of pDC or genetic deficiencies for MyD88 or TLR9 drastically decreased production of IFN-I, but not the protective antiviral responses. Moreover, MyD88, but not IFN-I receptor, deficiency could largely be compensated by Ly49H-mediated antiviral NK cell responses. Thus, contrary to the current dogma but consistent with the situation in humans, we conclude that, in mice, in our experimental settings, MyD88 is redundant for IFN-I responses and overall defense against a systemic herpes virus infection. Moreover, we identified direct NK cell sensing of infected cells as one mechanism able to compensate for MyD88 deficiency in mice. Similar mechanisms likely contribute to protect MyD88- or IRAK4-deficient patients from viral infections.
在小鼠中,浆细胞样树突状细胞(pDC)和自然杀伤(NK)细胞均有助于抵抗包括小鼠巨细胞病毒(MCMV)在内的疱疹病毒的全身感染。在MCMV感染期间,pDC是I型干扰素(IFN-I)的主要来源。这种反应需要Toll样受体7和9介导的pDC内在的MyD88依赖性信号传导。只要NK细胞表达适当的识别受体,如Ly49H,就可以直接感知并杀死MCMV感染的细胞。这些反应中任何一种的缺失都会增加感染的易感性。然而,这些抗病毒免疫反应的相对重要性以及它们之间的关系仍不清楚。在人类中,虽然IFN-I反应至关重要,但MyD88对于抗病毒免疫是可有可无的。因此,有人提出在人类自然感染期间,促进针对疱疹病毒全身感染的保护性免疫反应的机制具有更高的冗余性。有人假设但未得到证实,小鼠无法产生保护性的不依赖MyD88的IFN-I反应。在人类中,补偿MyD88缺陷的机制尚未阐明。为了解决这些问题,我们比较了MyD88、IFN-I受体和/或Ly49H缺陷的小鼠品系对MCMV感染的抵抗力和免疫反应。我们发现,选择性清除pDC或MyD88或TLR9的基因缺陷会大幅降低IFN-I的产生,但不会降低保护性抗病毒反应。此外,MyD88缺陷而非IFN-I受体缺陷在很大程度上可以由Ly49H介导的抗病毒NK细胞反应来补偿。因此,与当前的教条相反但与人类情况一致,我们得出结论,在小鼠中,在我们的实验环境中,MyD88对于IFN-I反应和针对全身性疱疹病毒感染的总体防御是多余的。此外,我们确定NK细胞对感染细胞的直接感知是一种能够补偿小鼠MyD88缺陷的机制。类似的机制可能有助于保护MyD88或IRAK4缺陷的患者免受病毒感染。