Tian Lei, Ding Sheng, You Yun, Li Tong-ruei, Liu Yan, Wu Xiaohui, Sun Ling, Xu Tian
State Key Laboratory of Genetic Engineering and National Center for International Research of Development and Disease, Fudan-Yale Center for Biomedical Research, Innovation Center for International Cooperation of Genetics and Development, Institute of Developmental Biology and Molecular Medicine, School of Life Sciences, Fudan University, Shanghai 200433, China Howard Hughes Medical Institute, Department of Genetics, Yale University School of Medicine, New Haven, CT 06536, USA.
Howard Hughes Medical Institute, Department of Genetics, Yale University School of Medicine, New Haven, CT 06536, USA.
Dis Model Mech. 2015 Jun;8(6):635-41. doi: 10.1242/dmm.019430. Epub 2015 Apr 16.
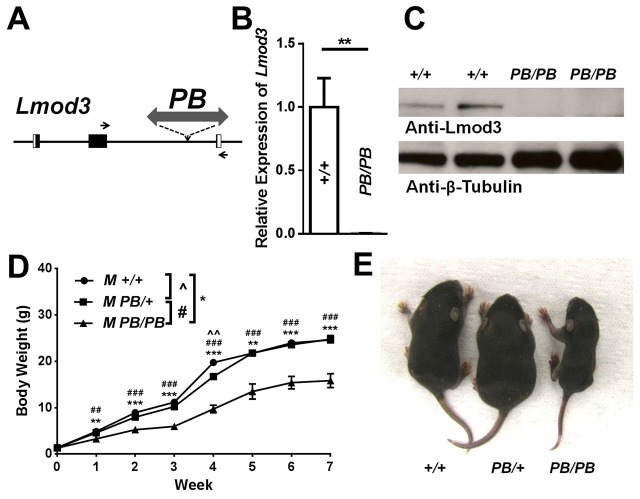
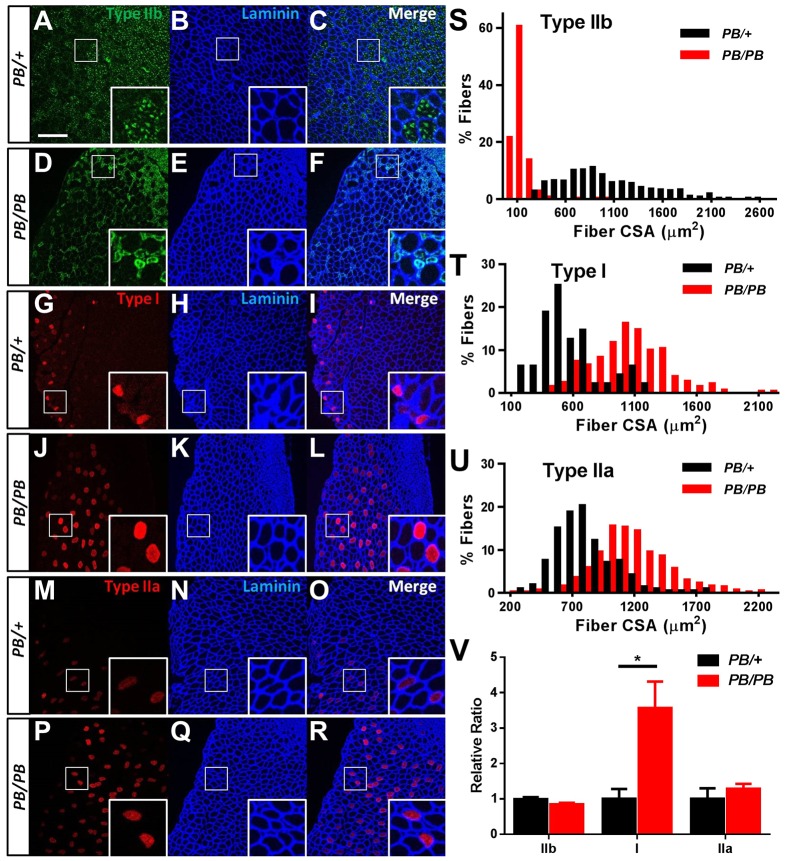
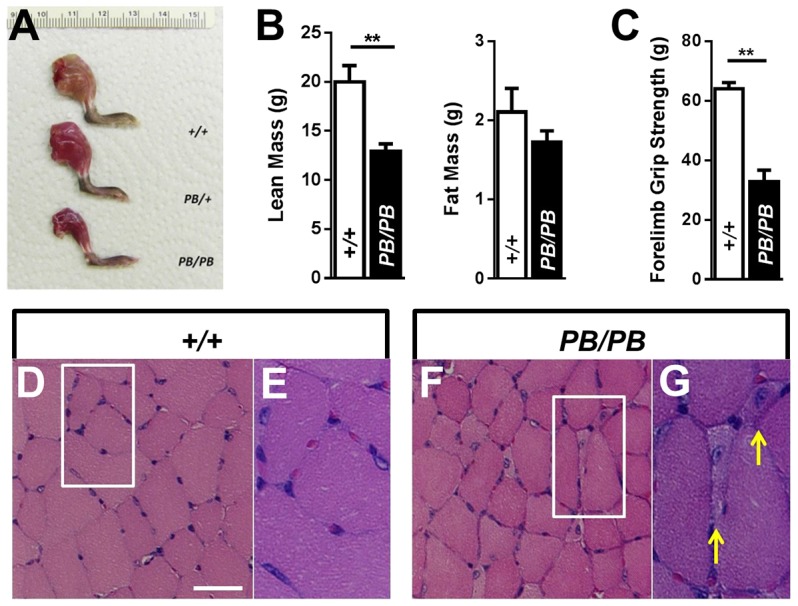
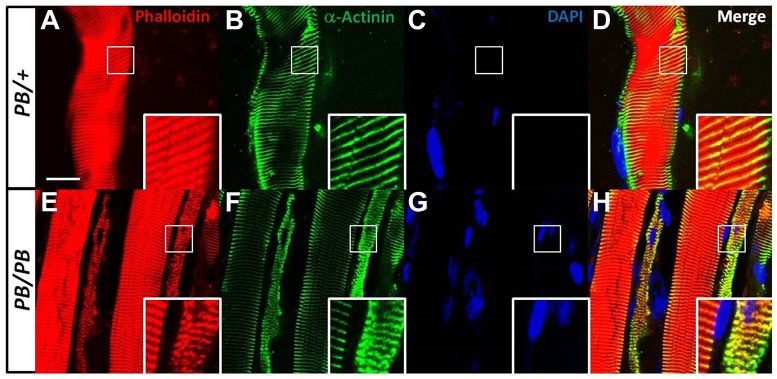
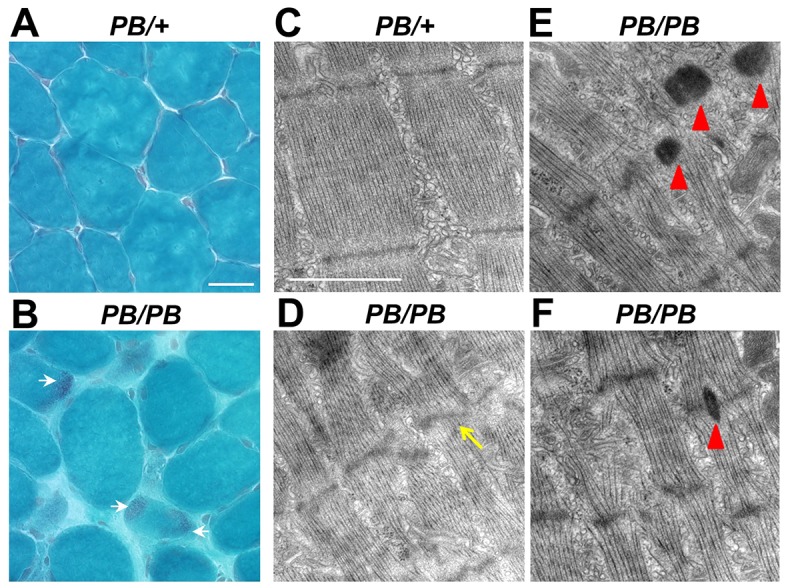
Nemaline myopathy (NM) is one of the most common forms of congenital myopathy, and affects either fast myofibers, slow myofibers, or both. However, an animal model for congenital myopathy with fast-myofiber-specific atrophy is not available. Furthermore, mutations in the leiomodin-3 (LMOD3) gene have recently been identified in a group of individuals with NM. However, it is not clear how loss of LMOD3 leads to NM. Here, we report a mouse mutant in which the piggyBac (PB) transposon is inserted into the Lmod3 gene and disrupts its expression. Lmod3(PB/PB) mice show severe muscle weakness and postnatal growth retardation. Electron microscopy and immunofluorescence studies of the mutant skeletal muscles revealed the presence of nemaline bodies, a hallmark of NM, and disorganized sarcomeric structures. Interestingly, Lmod3 deficiency caused muscle atrophy specific to the fast fibers. Together, our results show that Lmod3 is required in the fast fibers for sarcomere integrity, and this study offers the first NM mouse model with muscle atrophy that is specific to fast fibers. This model could be a valuable resource for interrogating myopathy pathogenesis and developing therapeutics for NM as well as other pathophysiological conditions with preferential atrophy of fast fibers, including cancer cachexia and sarcopenia.
杆状体肌病(NM)是先天性肌病最常见的形式之一,可影响快肌纤维、慢肌纤维或两者。然而,目前尚无针对伴有快肌纤维特异性萎缩的先天性肌病的动物模型。此外,最近在一组杆状体肌病患者中发现了平滑肌瘤素-3(LMOD3)基因的突变。然而,尚不清楚LMOD3的缺失如何导致杆状体肌病。在此,我们报告了一种小鼠突变体,其中猪尾巴(PB)转座子插入Lmod3基因并破坏其表达。Lmod3(PB/PB)小鼠表现出严重的肌肉无力和出生后生长迟缓。对突变体骨骼肌的电子显微镜和免疫荧光研究显示存在杆状体,这是杆状体肌病的一个标志,以及肌节结构紊乱。有趣的是,Lmod3缺乏导致快纤维特异性肌肉萎缩。总之,我们的结果表明,快纤维中Lmod3对于肌节完整性是必需的,并且本研究提供了首个具有快纤维特异性肌肉萎缩的杆状体肌病小鼠模型。该模型可能是用于探究肌病发病机制以及开发针对杆状体肌病和其他具有快纤维优先萎缩的病理生理状况(包括癌症恶病质和肌肉减少症)的治疗方法的宝贵资源。