Division of Pediatric Hematology/Oncology, Department of Pediatrics and Adolescent Medicine, Medical University Graz , Graz , Austria.
Division of Pediatric Hematology/Oncology, Department of Pediatrics, Technische Universität München , Munich , Germany.
Front Pediatr. 2015 Jun 8;3:50. doi: 10.3389/fped.2015.00050. eCollection 2015.
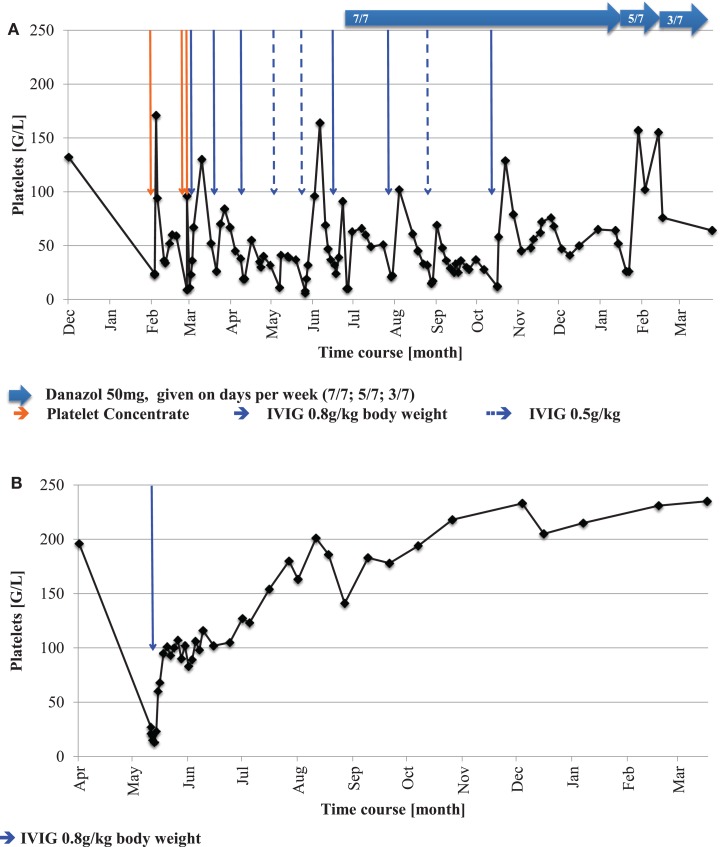
Thrombocytopenia and pancytopenia, occurring in patients with Fanconi anemia (FA), are interpreted either as progression to bone marrow failure or as developing myelodysplasia. On the other hand, immune thrombocytopenia (ITP) represents an acquired and often self-limiting benign hematologic disorder, associated with peripheral, immune-mediated, platelet destruction requiring different management modalities than those used in congenital bone marrow failure syndromes, including FA. Here, we describe the clinical course of two independent FA patients with atypical - namely immune - thrombocytopenia. While in one patient belonging to complementation group FA-A, the ITP started at 17 months of age and showed a chronically persisting course with severe purpura, responding well to intravenous immunoglobulins (IVIG) and later also danazol, a synthetic androgen, the other patient (of complementation group FA-D2) had a self-limiting course that resolved after one administration of IVIG. No cytogenetic aberrations or bone marrow abnormalities other than FA-typical mild dysplasia were detected. Our data show that acute and chronic ITP may occur in FA patients and impose individual diagnostic and therapeutic challenges in this rare congenital bone marrow failure/tumor predisposition syndrome. The management and a potential context of immune pathogenesis with the underlying marrow disorder are discussed.
血小板减少症和全血细胞减少症发生在范可尼贫血(FA)患者中,要么被解释为骨髓衰竭的进展,要么被解释为骨髓增生异常的发生。另一方面,免疫性血小板减少症(ITP)代表一种获得性且常常是自限性的良性血液疾病,与外周免疫介导的血小板破坏有关,需要与包括 FA 在内的先天性骨髓衰竭综合征不同的治疗方式。在这里,我们描述了两位独立的 FA 患者的临床病程,他们患有非典型的免疫性血小板减少症。在属于 FA-A 补体组的一名患者中,ITP 于 17 个月大时开始出现,呈慢性持续病程,伴有严重的紫癜,对静脉注射免疫球蛋白(IVIG)和后来的合成雄激素达那唑反应良好,而另一名患者(属于 FA-D2 补体组)则有自限性病程,在一次 IVIG 治疗后得到缓解。除了 FA 典型的轻度发育不良外,未检测到其他细胞遗传学异常或骨髓异常。我们的数据表明,急性和慢性 ITP 可能发生在 FA 患者中,并在这种罕见的先天性骨髓衰竭/肿瘤易感性综合征中带来个体诊断和治疗挑战。讨论了管理和潜在的免疫发病机制以及潜在的骨髓疾病背景。