Driebe Elizabeth M, Sahl Jason W, Roe Chandler, Bowers Jolene R, Schupp James M, Gillece John D, Kelley Erin, Price Lance B, Pearson Talima R, Hepp Crystal M, Brzoska Pius M, Cummings Craig A, Furtado Manohar R, Andersen Paal S, Stegger Marc, Engelthaler David M, Keim Paul S
Pathogen Genomics Division, The Translational Genomics Research Institute, Flagstaff, Arizona, United States of America.
Center for Microbial Genetics and Genomics, Northern Arizona University, Flagstaff, Arizona, United States of America.
PLoS One. 2015 Jul 10;10(7):e0130955. doi: 10.1371/journal.pone.0130955. eCollection 2015.
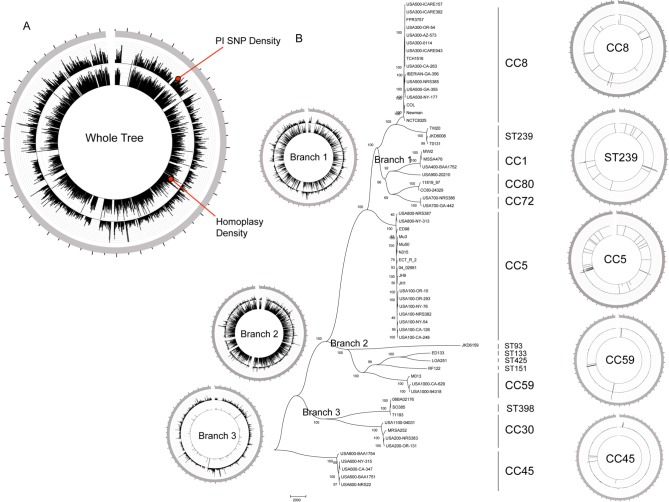
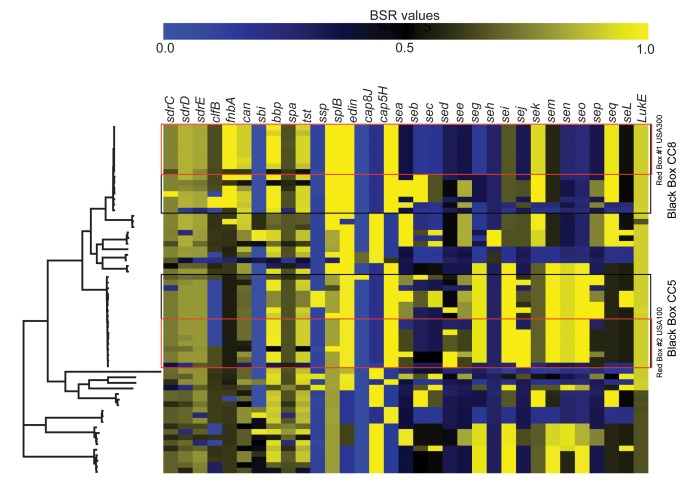
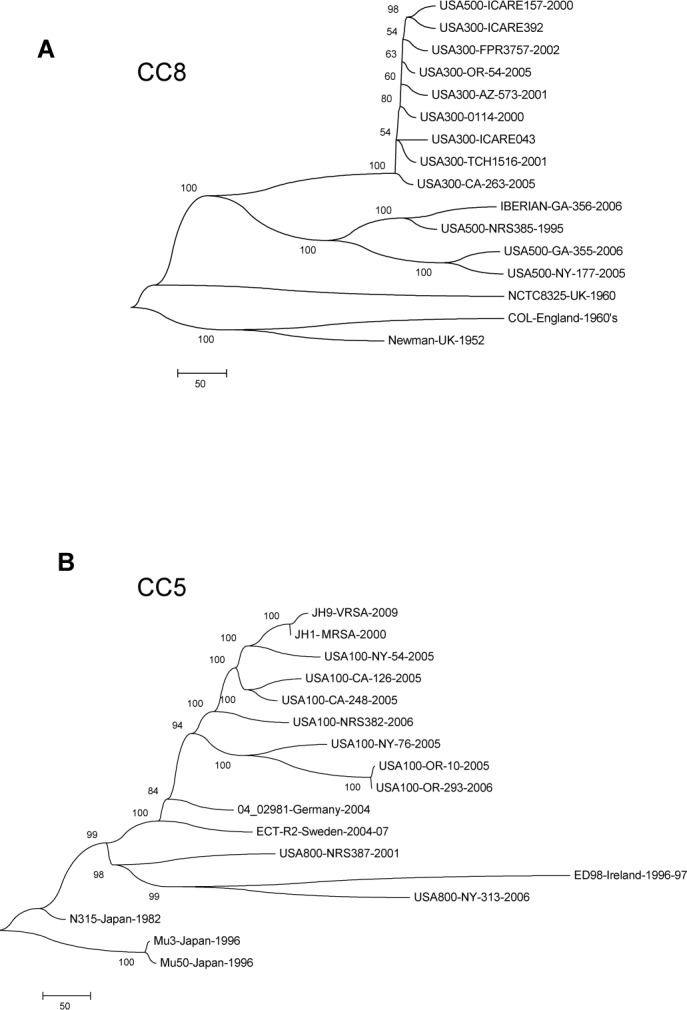
Staphylococcus aureus is an important clinical pathogen worldwide and understanding this organism's phylogeny and, in particular, the role of recombination, is important both to understand the overall spread of virulent lineages and to characterize outbreaks. To further elucidate the phylogeny of S. aureus, 35 diverse strains were sequenced using whole genome sequencing. In addition, 29 publicly available whole genome sequences were included to create a single nucleotide polymorphism (SNP)-based phylogenetic tree encompassing 11 distinct lineages. All strains of a particular sequence type fell into the same clade with clear groupings of the major clonal complexes of CC8, CC5, CC30, CC45 and CC1. Using a novel analysis method, we plotted the homoplasy density and SNP density across the whole genome and found evidence of recombination throughout the entire chromosome, but when we examined individual clonal lineages we found very little recombination. However, when we analyzed three branches of multiple lineages, we saw intermediate and differing levels of recombination between them. These data demonstrate that in S. aureus, recombination occurs across major lineages that subsequently expand in a clonal manner. Estimated mutation rates for the CC8 and CC5 lineages were different from each other. While the CC8 lineage rate was similar to previous studies, the CC5 lineage was 100-fold greater. Fifty known virulence genes were screened in all genomes in silico to determine their distribution across major clades. Thirty-three genes were present variably across clades, most of which were not constrained by ancestry, indicating horizontal gene transfer or gene loss.
金黄色葡萄球菌是全球重要的临床病原体,了解该菌的系统发育,尤其是重组的作用,对于理解致病谱系的整体传播以及确定疫情特征都非常重要。为进一步阐明金黄色葡萄球菌的系统发育,我们使用全基因组测序对35个不同菌株进行了测序。此外,还纳入了29个公开的全基因组序列,以构建包含11个不同谱系的基于单核苷酸多态性(SNP)的系统发育树。特定序列类型的所有菌株都落入同一进化枝,其中CC8、CC5、CC30、CC45和CC1等主要克隆复合体有明显的分组。使用一种新的分析方法,我们绘制了全基因组的同塑性密度和SNP密度,发现整个染色体都有重组的证据,但在检查单个克隆谱系时,我们发现重组很少。然而,当我们分析多个谱系的三个分支时,发现它们之间存在中等程度且不同水平的重组。这些数据表明,在金黄色葡萄球菌中,重组发生在主要谱系之间,随后以克隆方式扩展。CC8和CC5谱系的估计突变率彼此不同。虽然CC8谱系的速率与先前研究相似,但CC5谱系的速率高出100倍。在所有基因组中通过计算机筛选了50个已知的毒力基因,以确定它们在主要进化枝中的分布。33个基因在进化枝中存在差异,其中大多数不受祖先限制,表明存在水平基因转移或基因丢失。