Ratbi Ilham, Lyahyai Jaber, Kabiri Meryem, Banouar Meryem, Zerkaoui Maria, Barkat Amina, Sefiania Abdelaziz
Ilham Ratbi, Département de Génétique Médicale,, Institut National d'Hygiène, 27, Avenue Ibn Batouta,, B.P. 769 Rabat - Morocco, T: (+212) 613 58 67 97, F: (+212) 537 77 20 67,
Ann Saudi Med. 2015 Mar-Apr;35(2):170-2. doi: 10.5144/0256-4947.2015.170.
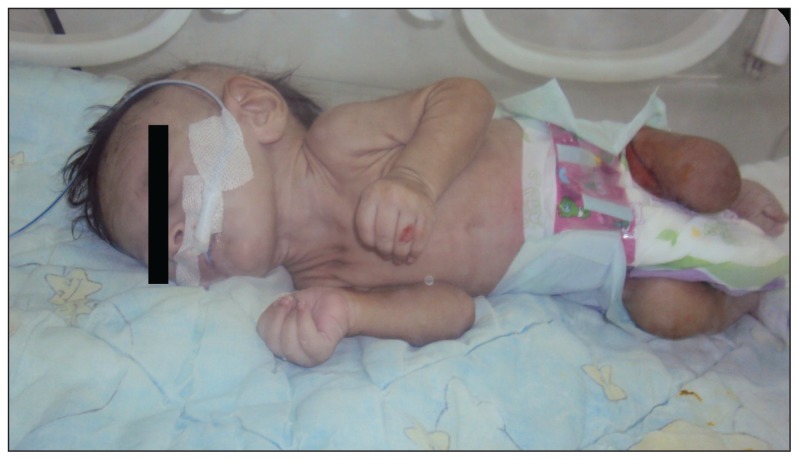
Sanjad-Sakati syndrome (SSS) or hypoparathyroidism-retardation-dysmorphism syndrome (HDR) is a rare autosomal recessive disorder. It is characterized by the association of congenital hypothyroidism, growth retardation, psychomotor retardation, epilepsy, dysmorphic features (microcephaly, facial, eye, and dental anomalies), and abnormalities of the extremities. The prevalence of SSS is unknown. Reported patients are were almost exclusively from the Arabian Peninsula. They are were all homozygous for the ancestral mutation c.155- 166del of the TBCE gene, also known as "the Bedouin mutation." We report on the first clinical and molecular description of a Moroccan patient with Sanjad-Sakati syndrome. He is was a carrier for the Bedouin mutation, not surprising, knowing that part of the Moroccan population has Arabian origin. This diagnosis allowed us to provide an appropriate management to the patient, to make a genetic counseling to his family, and to enrich genetic data on the Moroccan population.
桑贾德 - 萨卡蒂综合征(SSS)或甲状旁腺功能减退 - 发育迟缓 - 畸形综合征(HDR)是一种罕见的常染色体隐性疾病。其特征为先天性甲状腺功能减退、生长发育迟缓、精神运动发育迟缓、癫痫、畸形特征(小头畸形、面部、眼睛和牙齿异常)以及四肢异常。SSS的患病率尚不清楚。报道的患者几乎都来自阿拉伯半岛。他们均为TBCE基因祖传突变c.155 - 166del的纯合子,该突变也被称为“贝都因突变”。我们报告了首例摩洛哥桑贾德 - 萨卡蒂综合征患者的临床和分子描述。他是贝都因突变的携带者,鉴于部分摩洛哥人口有阿拉伯血统,这并不奇怪。该诊断使我们能够为患者提供适当的治疗,为其家人进行遗传咨询,并丰富摩洛哥人群的遗传数据。