Kishore Kamal, de Pretis Stefano, Lister Ryan, Morelli Marco J, Bianchi Valerio, Amati Bruno, Ecker Joseph R, Pelizzola Mattia
Center for Genomic Science of IIT@SEMM, Istituto Italiano di Tecnologia (IIT), Milano, 20139, Italy.
Australian Research Council Centre of Excellence in Plant Energy Biology, The University of Western Australia, Perth, WA, 6009, Australia.
BMC Bioinformatics. 2015 Sep 29;16:313. doi: 10.1186/s12859-015-0742-6.
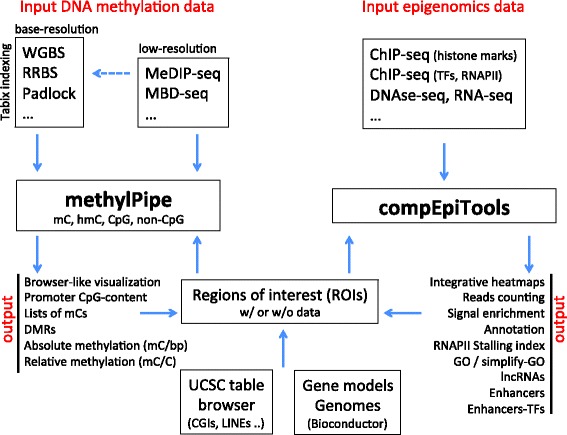
Numerous methods are available to profile several epigenetic marks, providing data with different genome coverage and resolution. Large epigenomic datasets are then generated, and often combined with other high-throughput data, including RNA-seq, ChIP-seq for transcription factors (TFs) binding and DNase-seq experiments. Despite the numerous computational tools covering specific steps in the analysis of large-scale epigenomics data, comprehensive software solutions for their integrative analysis are still missing. Multiple tools must be identified and combined to jointly analyze histone marks, TFs binding and other -omics data together with DNA methylation data, complicating the analysis of these data and their integration with publicly available datasets.
To overcome the burden of integrating various data types with multiple tools, we developed two companion R/Bioconductor packages. The former, methylPipe, is tailored to the analysis of high- or low-resolution DNA methylomes in several species, accommodating (hydroxy-)methyl-cytosines in both CpG and non-CpG sequence context. The analysis of multiple whole-genome bisulfite sequencing experiments is supported, while maintaining the ability of integrating targeted genomic data. The latter, compEpiTools, seamlessly incorporates the results obtained with methylPipe and supports their integration with other epigenomics data. It provides a number of methods to score these data in regions of interest, leading to the identification of enhancers, lncRNAs, and RNAPII stalling/elongation dynamics. Moreover, it allows a fast and comprehensive annotation of the resulting genomic regions, and the association of the corresponding genes with non-redundant GeneOntology terms. Finally, the package includes a flexible method based on heatmaps for the integration of various data types, combining annotation tracks with continuous or categorical data tracks.
methylPipe and compEpiTools provide a comprehensive Bioconductor-compliant solution for the integrative analysis of heterogeneous epigenomics data. These packages are instrumental in providing biologists with minimal R skills a complete toolkit facilitating the analysis of their own data, or in accelerating the analyses performed by more experienced bioinformaticians.
有多种方法可用于分析多种表观遗传标记,从而提供具有不同基因组覆盖范围和分辨率的数据。随后会生成大量表观基因组数据集,并且这些数据集通常会与其他高通量数据相结合,包括RNA测序、用于转录因子(TFs)结合的ChIP测序以及DNase测序实验。尽管有众多计算工具可涵盖大规模表观基因组学数据分析中的特定步骤,但仍缺少用于其综合分析的全面软件解决方案。必须识别并组合多个工具,以便将组蛋白标记、TFs结合以及其他组学数据与DNA甲基化数据一起进行联合分析,这使得这些数据的分析及其与公开可用数据集的整合变得复杂。
为了克服使用多个工具整合各种数据类型的负担,我们开发了两个配套的R/Bioconductor软件包。前者methylPipe专门用于分析多个物种中的高分辨率或低分辨率DNA甲基化组,可处理CpG和非CpG序列背景下的(羟基 - )甲基化胞嘧啶。支持对多个全基因组亚硫酸氢盐测序实验进行分析,同时保持整合靶向基因组数据的能力。后者compEpiTools无缝整合了使用methylPipe获得的结果,并支持将其与其他表观基因组学数据进行整合。它提供了多种方法来对感兴趣区域中的这些数据进行评分,从而识别增强子、长链非编码RNA以及RNA聚合酶II的停顿/延伸动态。此外,它允许对所得基因组区域进行快速而全面的注释,并将相应基因与非冗余的基因本体术语相关联。最后,该软件包包括一种基于热图的灵活方法,用于整合各种数据类型,将注释轨迹与连续或分类数据轨迹相结合。
methylPipe和compEpiTools为异质表观基因组学数据的综合分析提供了一个全面的符合Bioconductor标准的解决方案。这些软件包有助于为具备最少R技能的生物学家提供一个完整的工具包,便于他们分析自己的数据,或者加速经验更丰富的生物信息学家所进行的分析。