Cantarella Concita, D'Agostino Nunzio
Consiglio per la ricerca in agricoltura e l'analisi dell'economia agraria - Centro di ricerca per l'orticoltura, Via Cavalleggeri 25, 84098, Pontecagnano Faiano, Italy.
BMC Res Notes. 2015 Oct 1;8:525. doi: 10.1186/s13104-015-1474-4.
With the advent of high-throughput sequencing technologies large-scale identification of microsatellites became affordable and was especially directed to non-model species. By contrast, few efforts have been published toward the automatic identification of polymorphic microsatellites by exploiting sequence redundancy. Few tools for genotyping microsatellite repeats have been implemented so far that are able to manage huge amount of sequence data and handle the SAM/BAM file format. Most of them have been developed for and tested on human or model organisms with high quality reference genomes.
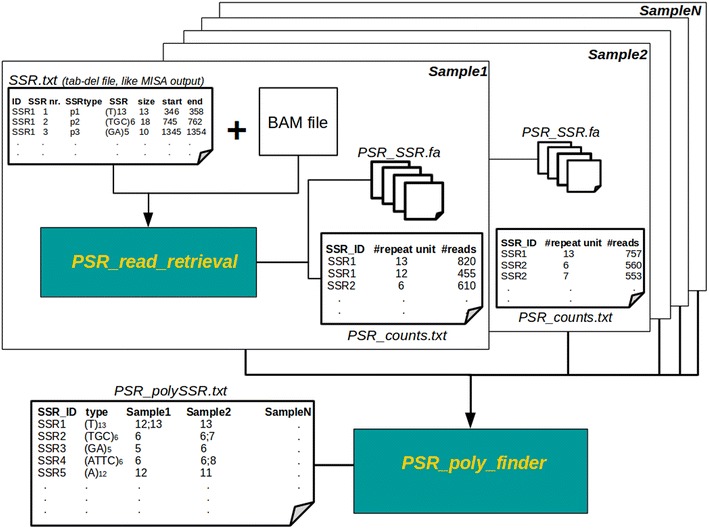
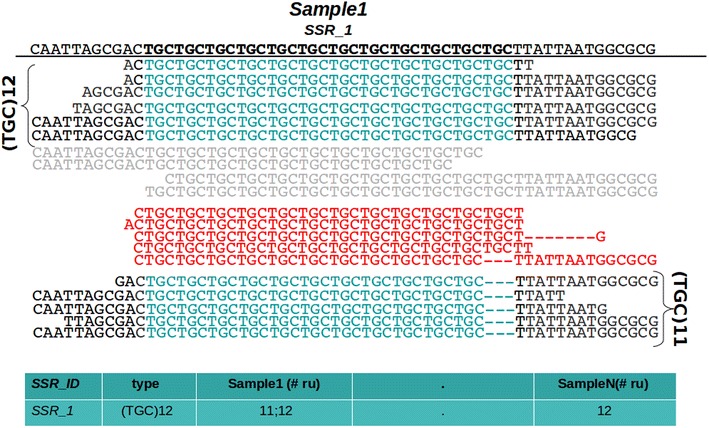
In this note we describe polymorphic SSR retrieval (PSR), a read counter and simple sequence repeat (SSR) length polymorphism detection tool. It is written in Perl and was developed to identify length polymorphisms in perfect microsatellites exploiting next generation sequencing (NGS) data. PSR has been developed bearing in mind plant non-model species for which de novo transcriptome assembly is generally the first sequence resource available to be used for SSR-mining. PSR is divided into two modules: the read-counting module (PSR_read_retrieval) identifies all the reads that cover the full-length of perfect microsatellites; the comparative module (PSR_poly_finder) detects both heterozygous and homozygous alleles at each microsatellite locus across all genotypes under investigation. Two threshold values to call a length polymorphism and reduce the number of false positives can be defined by the user: the minimum number of reads overlapping the repetitive stretch and the minimum read depth. The first parameter determines if the microsatellite-containing sequence must be processed or not, while the second one is decisive for the identification of minor alleles. PSR was tested on two different case studies. The first study aims at the identification of polymorphic SSRs in a set of de novo assembled transcripts defined by RNA-sequencing of two different plant genotypes. The second research activity aims to investigate sequence variations within a collection of newly sequenced chloroplast genomes. In both the cases PSR results are in agreement with those obtained by capillary gel separation.
PSR has been specifically developed from the need to automate the gene-based and genome-wide identification of polymorphic microsatellites from NGS data. It overcomes the limits related to the existing and time-consuming efforts based on tools developed in the pre-NGS era.
随着高通量测序技术的出现,大规模鉴定微卫星变得可行,尤其适用于非模式物种。相比之下,利用序列冗余自动鉴定多态性微卫星的研究较少。目前,很少有能够处理大量序列数据并处理SAM/BAM文件格式的微卫星重复基因分型工具。它们中的大多数是针对具有高质量参考基因组的人类或模式生物开发并进行测试的。
在本报告中,我们描述了多态性SSR检索工具(PSR),这是一种读取计数器和简单序列重复(SSR)长度多态性检测工具。它用Perl编写,旨在利用下一代测序(NGS)数据识别完美微卫星中的长度多态性。PSR的开发考虑到了植物非模式物种,对于这些物种而言,从头转录组组装通常是可用于SSR挖掘的首个序列资源。PSR分为两个模块:读取计数模块(PSR_read_retrieval)识别覆盖完美微卫星全长的所有读取;比较模块(PSR_poly_finder)检测所有被研究基因型中每个微卫星位点的杂合和纯合等位基因。用户可以定义两个阈值来判定长度多态性并减少假阳性数量:与重复片段重叠的最小读取数和最小读取深度。第一个参数决定是否必须处理包含微卫星的序列,而第二个参数对于次要等位基因的识别起决定性作用。PSR在两个不同的案例研究中进行了测试。第一个研究旨在通过对两种不同植物基因型进行RNA测序来鉴定一组从头组装转录本中的多态性SSR。第二项研究活动旨在研究一组新测序的叶绿体基因组中的序列变异。在这两种情况下,PSR的结果都与通过毛细管凝胶分离获得的结果一致。
PSR是根据从NGS数据中自动进行基于基因和全基因组的多态性微卫星鉴定的需求而专门开发的。它克服了与基于NGS时代之前开发的工具的现有且耗时的工作相关的限制。