Gacesa Ranko, Chung Ray, Dunn Simon R, Weston Andrew J, Jaimes-Becerra Adrian, Marques Antonio C, Morandini André C, Hranueli Daslav, Starcevic Antonio, Ward Malcolm, Long Paul F
Institute of Pharmaceutical Science, King's College London, 150 Stamford Street, London, SE1 9NH, UK.
Proteomics Facility, Institute of Psychiatry, Psychology & Neuroscience, King's College London, 16 De Crespigny Park, London, SE5 8AF, UK.
BMC Genomics. 2015 Oct 13;16:774. doi: 10.1186/s12864-015-1976-4.
Gene duplication followed by adaptive selection is a well-accepted process leading to toxin diversification in venoms. However, emergent genomic, transcriptomic and proteomic evidence now challenges this role to be at best equivocal to other processess . Cnidaria are arguably the most ancient phylum of the extant metazoa that are venomous and such provide a definitive ancestral anchor to examine the evolution of this trait.
Here we compare predicted toxins from the translated genome of the coral Acropora digitifera to putative toxins revealed by proteomic analysis of soluble proteins discharged from nematocysts, to determine the extent to which gene duplications contribute to venom innovation in this reef-building coral species. A new bioinformatics tool called HHCompare was developed to detect potential gene duplications in the genomic data, which is made freely available ( https://github.com/rgacesa/HHCompare ).
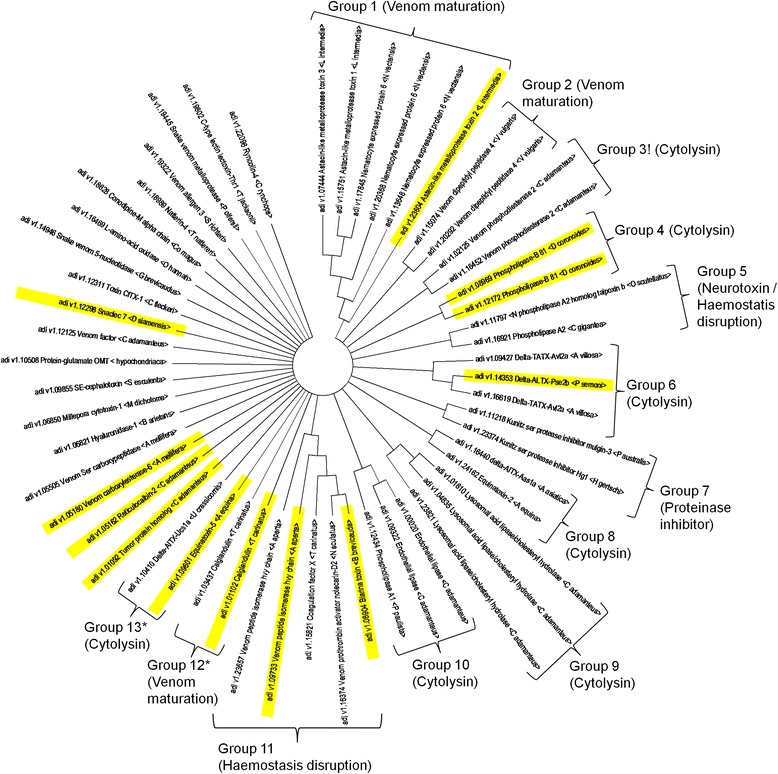
A total of 55 potential toxin encoding genes could be predicted from the A. digitifera genome, of which 36 (65 %) had likely arisen by gene duplication as evinced using the HHCompare tool and verified using two standard phylogeny methods. Surprisingly, only 22 % (12/55) of the potential toxin repertoire could be detected following rigorous proteomic analysis, for which only half (6/12) of the toxin proteome could be accounted for as peptides encoded by the gene duplicates. Biological activities of these toxins are dominatedby putative phospholipases and toxic peptidases.
Gene expansions in A. digitifera venom are the most extensive yet described in any venomous animal, and gene duplication plays a significant role leading to toxin diversification in this coral species. Since such low numbers of toxins were detected in the proteome, it is unlikely that the venom is evolving rapidly by prey-driven positive natural selection. Rather we contend that the venom has a defensive role deterring predation or harm from interspecific competition and overgrowth by fouling organisms. Factors influencing translation of toxin encoding genes perhaps warrants more profound experimental consideration.
基因复制后进行适应性选择是一个被广泛接受的过程,它导致毒液中毒素的多样化。然而,新出现的基因组学、转录组学和蛋白质组学证据现在对这一作用提出了挑战,认为其至多与其他过程一样模棱两可。刺胞动物门可以说是现存有毒后生动物中最古老的门类,因此为研究这一特征的进化提供了一个明确的祖先锚点。
在这里,我们将珊瑚鹿角珊瑚的翻译基因组中预测的毒素与通过对刺丝囊中排出的可溶性蛋白质进行蛋白质组分析揭示的假定毒素进行比较,以确定基因复制在这种造礁珊瑚物种的毒液创新中所起的作用程度。开发了一种名为HHCompare的新生物信息学工具来检测基因组数据中的潜在基因复制,该工具可免费获取(https://github.com/rgacesa/HHCompare)。
从鹿角珊瑚基因组中总共可以预测出55个潜在的毒素编码基因,其中36个(65%)可能是通过基因复制产生的,这是使用HHCompare工具证明并通过两种标准系统发育方法验证的。令人惊讶的是,经过严格的蛋白质组分析后,只能检测到潜在毒素库的22%(12/55),其中毒素蛋白质组中只有一半(6/12)可以被解释为由基因复制体编码的肽。这些毒素的生物活性主要由假定的磷脂酶和有毒肽酶主导。
鹿角珊瑚毒液中的基因扩增是迄今为止在任何有毒动物中描述的最广泛的,基因复制在导致这种珊瑚物种毒素多样化方面发挥了重要作用。由于在蛋白质组中检测到的毒素数量如此之少,毒液不太可能通过猎物驱动的正向自然选择快速进化。相反,我们认为毒液具有防御作用,可阻止捕食或来自种间竞争以及污损生物过度生长造成的伤害。影响毒素编码基因翻译的因素可能值得更深入的实验考虑。