Pastula Daniel M, Burish Mark, Reis Gerald F, Bollen Andrew, Cha Soonmee, Ralph Jeffrey, Douglas Vanja C
Department of Neurology, UCSF Medical Center, Box 0114, 505 Parnassus Ave, M798, San Francisco, CA, 94143-0114, USA.
Department of Anatomic Pathology, University of California San Francisco Medical School, San Francisco, CA, USA.
BMC Neurol. 2015 Oct 14;15:203. doi: 10.1186/s12883-015-0470-6.
Hemophagocytic lymphohistiocytosis (HLH) is a clinical syndrome with both genetic and acquired causes characterized by elevated cytokine levels, hyperinflammation, and overactivation of lymphocytes and macrophages. It is typically a systemic disease with variable degrees of CNS involvement. Cases with predominantly central nervous system (CNS) involvement are very rare, with the vast majority of these occurring in infants and young children. This report documents a case of adult-onset CNS-HLH involving a middle-aged man.
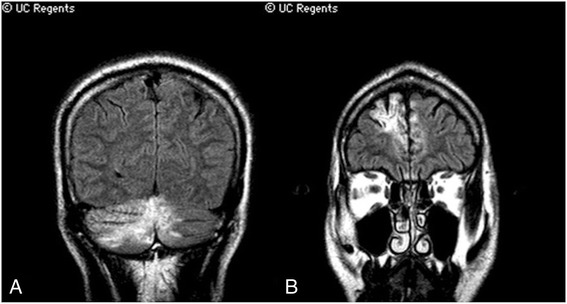
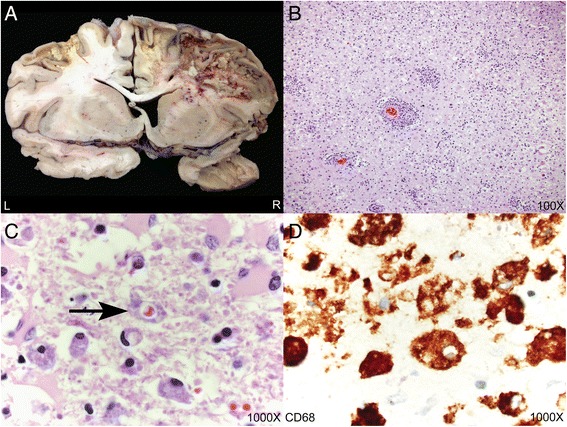
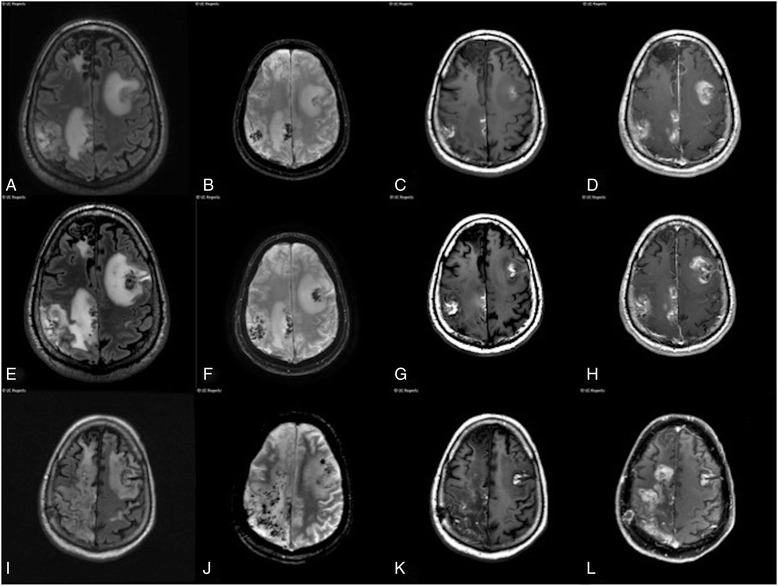
A 55 year-old man developed progressive left hemiparesis and aphasia over the course of several months. Brain MRI showed multifocal, mass-like enhancing lesions with increased susceptibility consistent with blood products. An extensive workup for infectious, autoimmune, and neoplastic etiologies was significant only for a markedly elevated serum ferritin at 1456 ng/mL. Two brain biopsies showed a non-specific inflammatory process. The patient was treated empirically with steroids and plasmapheresis, but he continued to suffer a progressive neurological decline and died one year after onset of neurological symptoms. Autopsy revealed profound histiocytic infiltration, perivascular lymphocytosis, and emperipolesis, compatible with CNS-HLH.
This case report describes an exceedingly rare presentation of an adult patient with CNS predominant HLH. This diagnosis should be considered in the differential diagnosis of adults presenting with progressive brain lesions, even in the absence of typical systemic signs of HLH.
噬血细胞性淋巴组织细胞增生症(HLH)是一种具有遗传和后天病因的临床综合征,其特征为细胞因子水平升高、炎症反应过度以及淋巴细胞和巨噬细胞过度活化。它通常是一种全身性疾病,伴有不同程度的中枢神经系统受累。主要累及中枢神经系统(CNS)的病例非常罕见,其中绝大多数发生在婴幼儿中。本报告记录了一例成年发病的中枢神经系统HLH病例,患者为一名中年男性。
一名55岁男性在数月内逐渐出现左侧偏瘫和失语。脑部MRI显示多灶性、肿块样强化病变,磁敏感增加,与血液产物相符。针对感染、自身免疫和肿瘤病因进行的广泛检查仅发现血清铁蛋白显著升高,达1456 ng/mL。两次脑活检显示为非特异性炎症过程。患者接受了经验性的类固醇和血浆置换治疗,但他仍持续出现进行性神经功能衰退,并在神经症状出现一年后死亡。尸检显示有大量组织细胞浸润、血管周围淋巴细胞增多和血细胞吞噬现象,符合中枢神经系统HLH。
本病例报告描述了一名成年患者以中枢神经系统为主的HLH极为罕见的表现。即使在没有HLH典型全身症状的情况下,对于出现进行性脑病变的成年人进行鉴别诊断时也应考虑这一诊断。