Narayanan Mohan, Ramsey Keri, Grebe Theresa, Schrauwen Isabelle, Szelinger Szabolcs, Huentelman Matthew, Craig David, Narayanan Vinodh
Arizona Pediatric Neurology & Neurogenetics Associates, Phoenix, AZ, USA ; Barrow Neurological Institute, Phoenix, AZ, USA.
Center for Rare Childhood Disorders, Translational Genomics Research Institute, Phoenix, AZ, USA ; Neurogenomics Division, Translational Genomics Research Institute, Phoenix, AZ, USA.
F1000Res. 2015 Sep 28;4:912. doi: 10.12688/f1000research.7106.1. eCollection 2015.
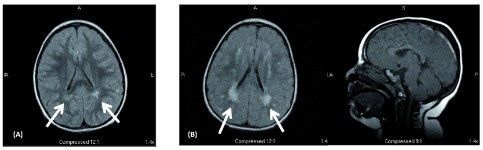
Microcephaly is a fairly common feature observed in children with delayed development, defined as head circumference less than 2 standard deviations below the mean for age and gender. It may be the result of an acquired insult to the brain, such prenatal or perinatal brain injury (congenital infection or hypoxic ischemic encephalopathy), or be a part of a genetic syndrome. There are over 1000 conditions listed in OMIM (Online Mendelian Inheritance in Man) where microcephaly is a key finding; many of these are associated with specific somatic features and non-CNS anomalies. The term primary microcephaly is used when microcephaly and delayed development are the primary features, and they are not part of another recognized syndrome. In this case report, we present the clinical features of siblings (brother and sister) with primary microcephaly and delayed development, and subtle dysmorphic features. Both children had brain MRI studies that showed periventricular and subcortical T2/FLAIR hyperintensities, without signs of white matter volume loss, and no parenchymal calcifications by CT scan. The family was enrolled in a research study for whole exome sequencing of probands and parents. Analysis of variants determined that the children were compound heterozygotes for nonsense mutations, c.277C>T (p.Arg93*) and c.397C>T (p.Arg133*), in the TRMT10A gene. Mutations in this gene have only recently been reported in children with microcephaly and early onset diabetes mellitus. Our report adds to current knowledge of TRMT10A related neurodevelopmental disorders and demonstrates imaging findings suggestive of delayed or abnormal myelination of the white matter in this disorder. Accurate diagnosis through genomic testing, as in the children described here, allows for early detection and management of medical complications, such as diabetes mellitus.
小头畸形是发育迟缓儿童中较为常见的特征,定义为头围低于年龄和性别的均值2个标准差以下。它可能是大脑后天受损的结果,如产前或围产期脑损伤(先天性感染或缺氧缺血性脑病),也可能是遗传综合征的一部分。在线人类孟德尔遗传数据库(OMIM)中列出了1000多种以小头畸形为关键发现的病症;其中许多与特定的躯体特征和非中枢神经系统异常有关。当小头畸形和发育迟缓是主要特征且不属于另一种公认的综合征时,使用原发性小头畸形这一术语。在本病例报告中,我们呈现了患有原发性小头畸形、发育迟缓及细微畸形特征的一对同胞(兄妹)的临床特征。两个孩子的脑部MRI检查均显示脑室周围和皮质下T2/FLAIR高信号,无白质体积减少的迹象,CT扫描未发现实质钙化。该家庭参与了一项针对先证者及其父母的全外显子测序研究。对变异的分析确定,这两个孩子是TRMT10A基因无义突变c.277C>T(p.Arg93*)和c.397C>T(p.Arg133*)的复合杂合子。该基因的突变最近才在患有小头畸形和早发性糖尿病的儿童中被报道。我们的报告增加了目前对TRMT10A相关神经发育障碍的认识,并展示了提示该疾病中白质髓鞘形成延迟或异常的影像学表现。如本文所述的儿童通过基因检测进行准确诊断,可以早期发现和管理糖尿病等医学并发症。