Tarighat S S, Santhanam R, Frankhouser D, Radomska H S, Lai H, Anghelina M, Wang H, Huang X, Alinari L, Walker A, Caligiuri M A, Croce C M, Li L, Garzon R, Li C, Baiocchi R A, Marcucci G
Molecular, Cellular, and Developmental Biology Graduate Program, The Ohio State University Comprehensive Cancer Center, Columbus, OH, USA.
Division of Hematology, Department of Medicine, The Ohio State University and The Ohio State University Comprehensive Cancer Center, Columbus, OH, USA.
Leukemia. 2016 Apr;30(4):789-99. doi: 10.1038/leu.2015.308. Epub 2015 Nov 5.
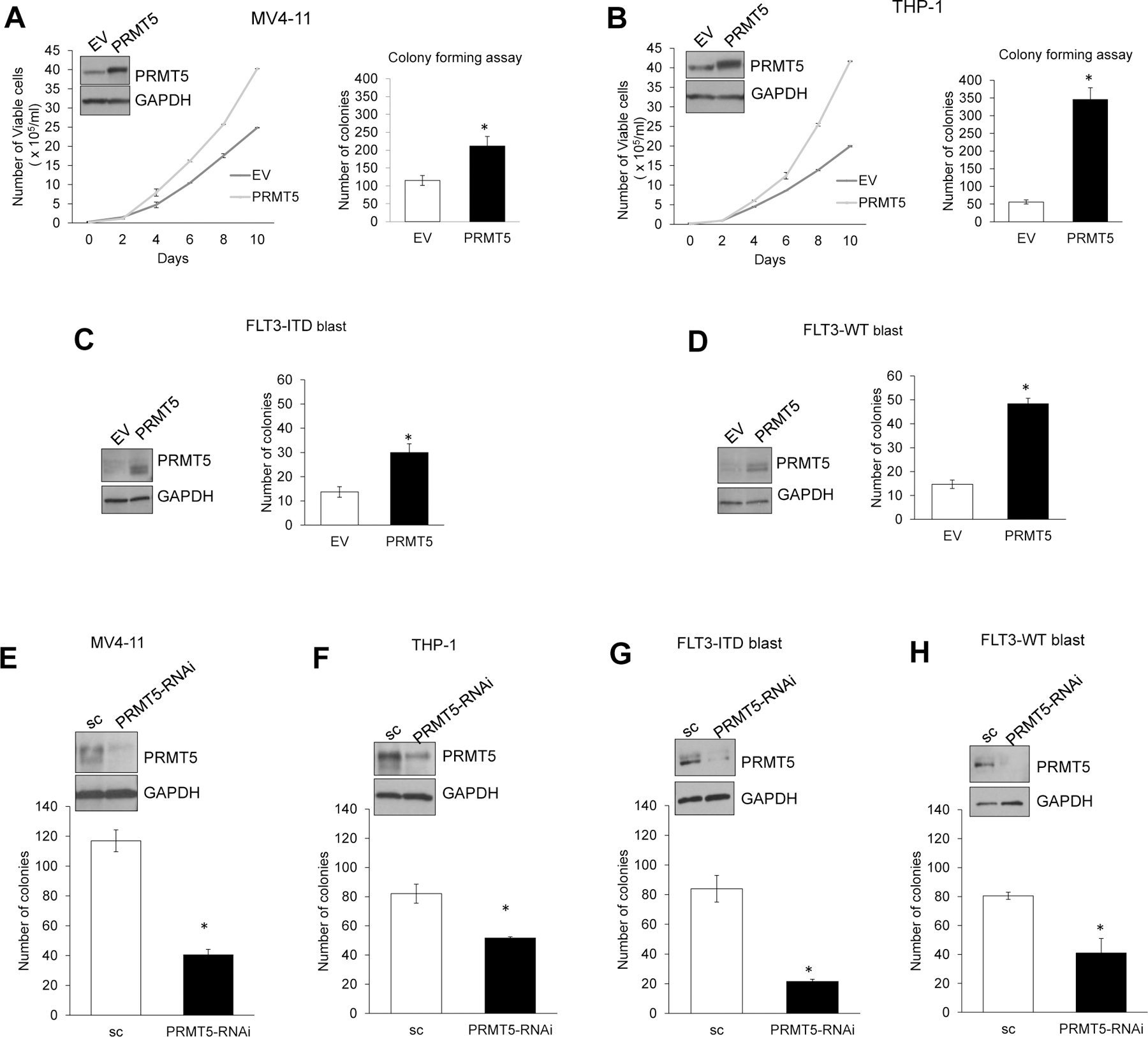
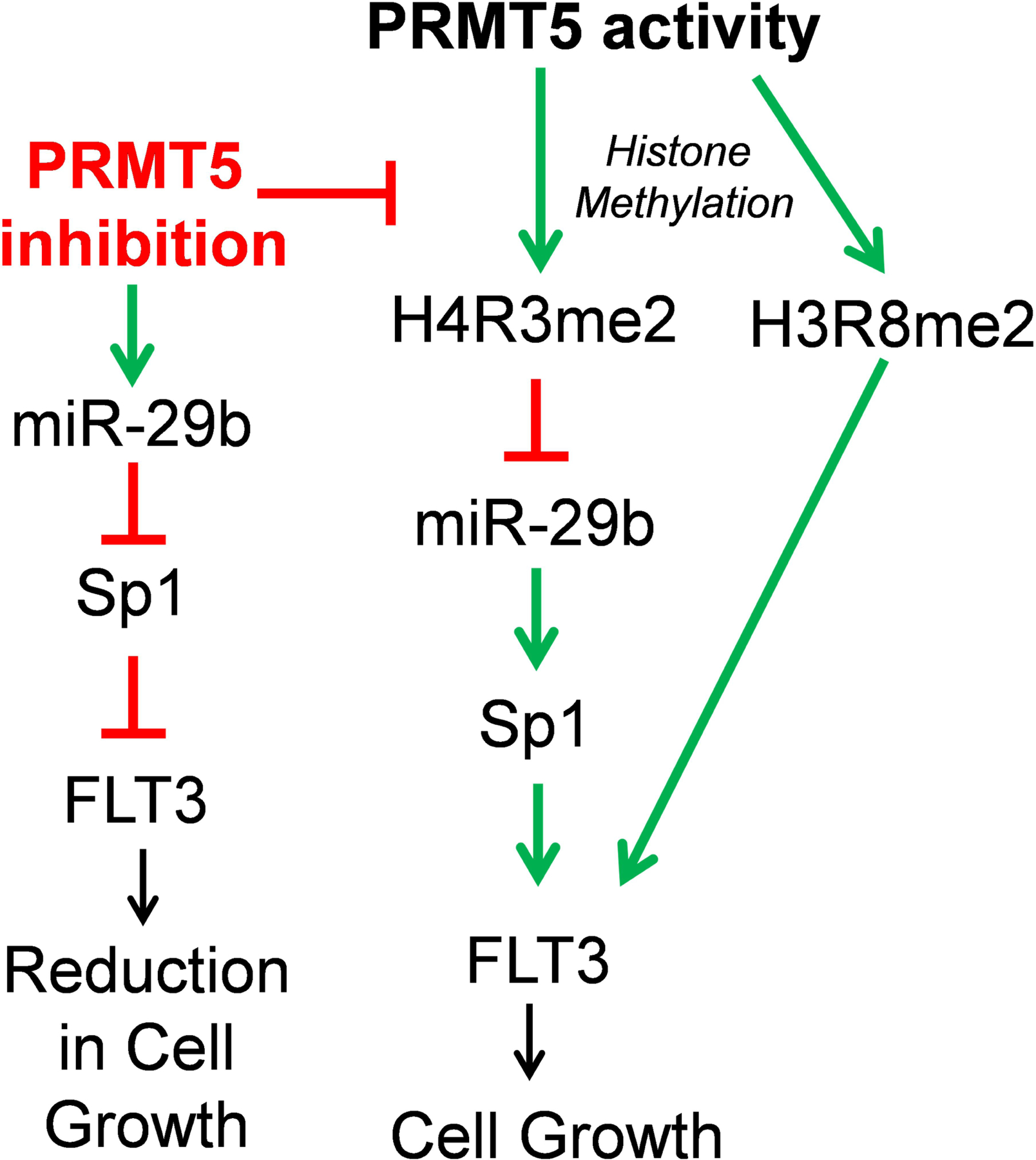
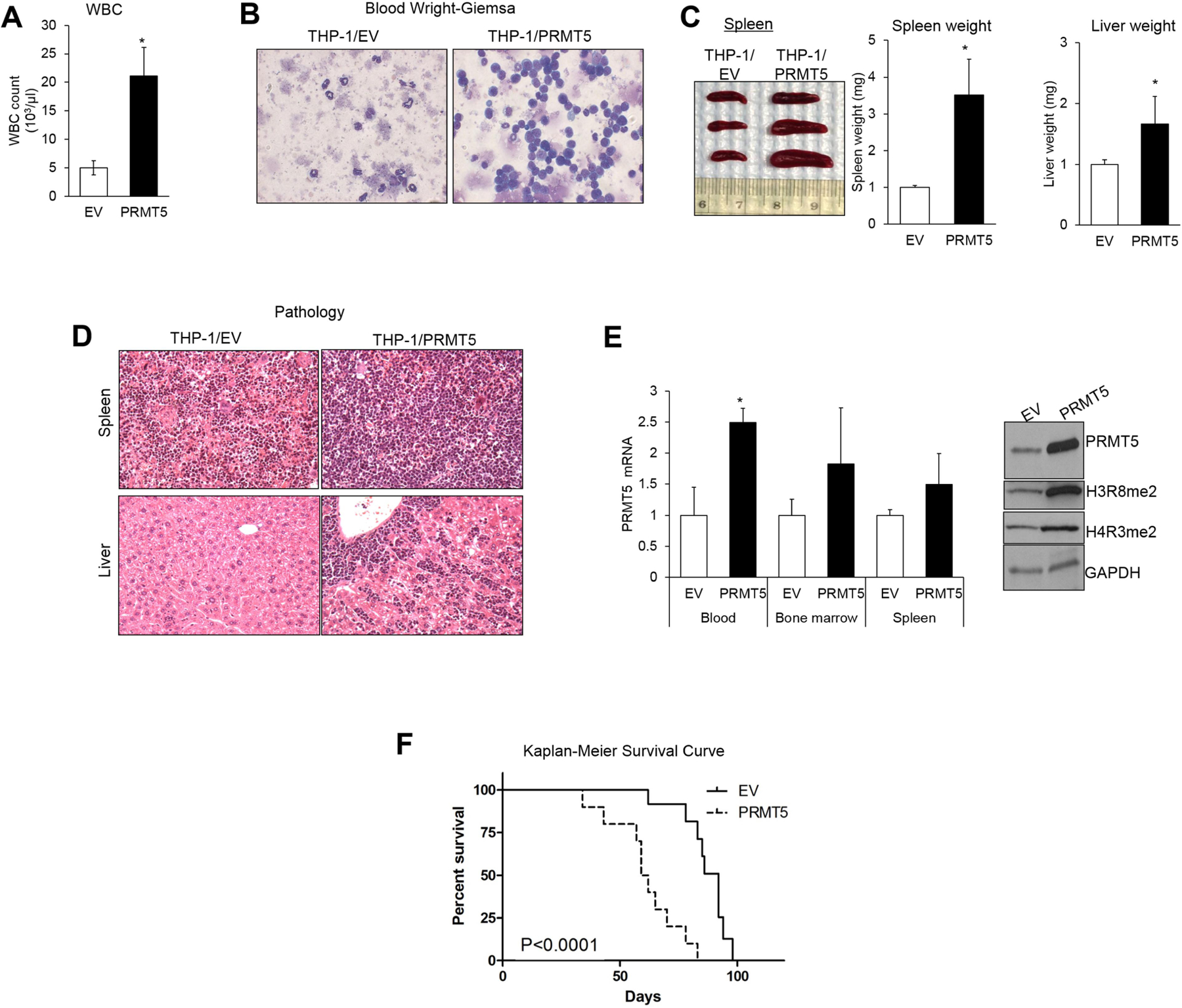
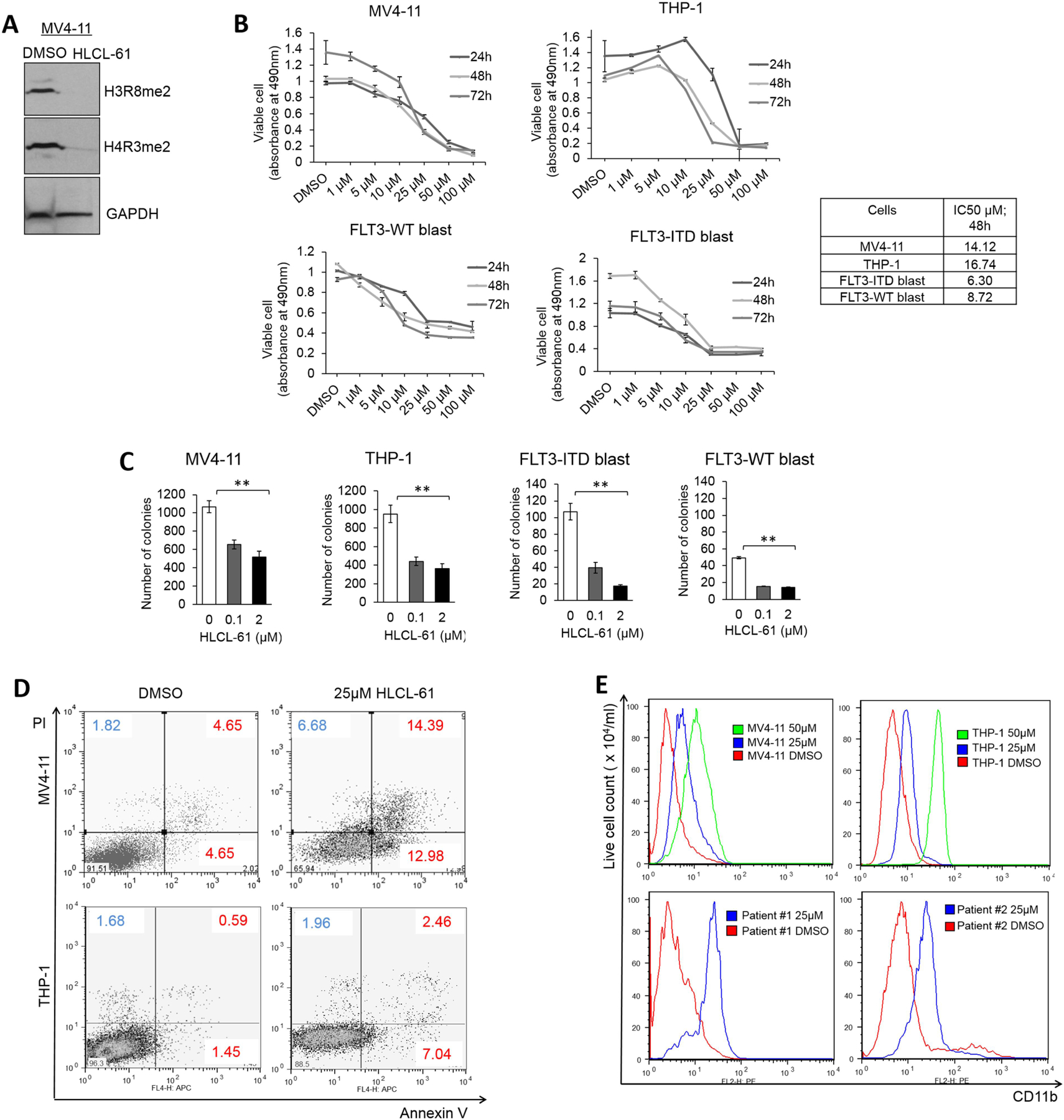
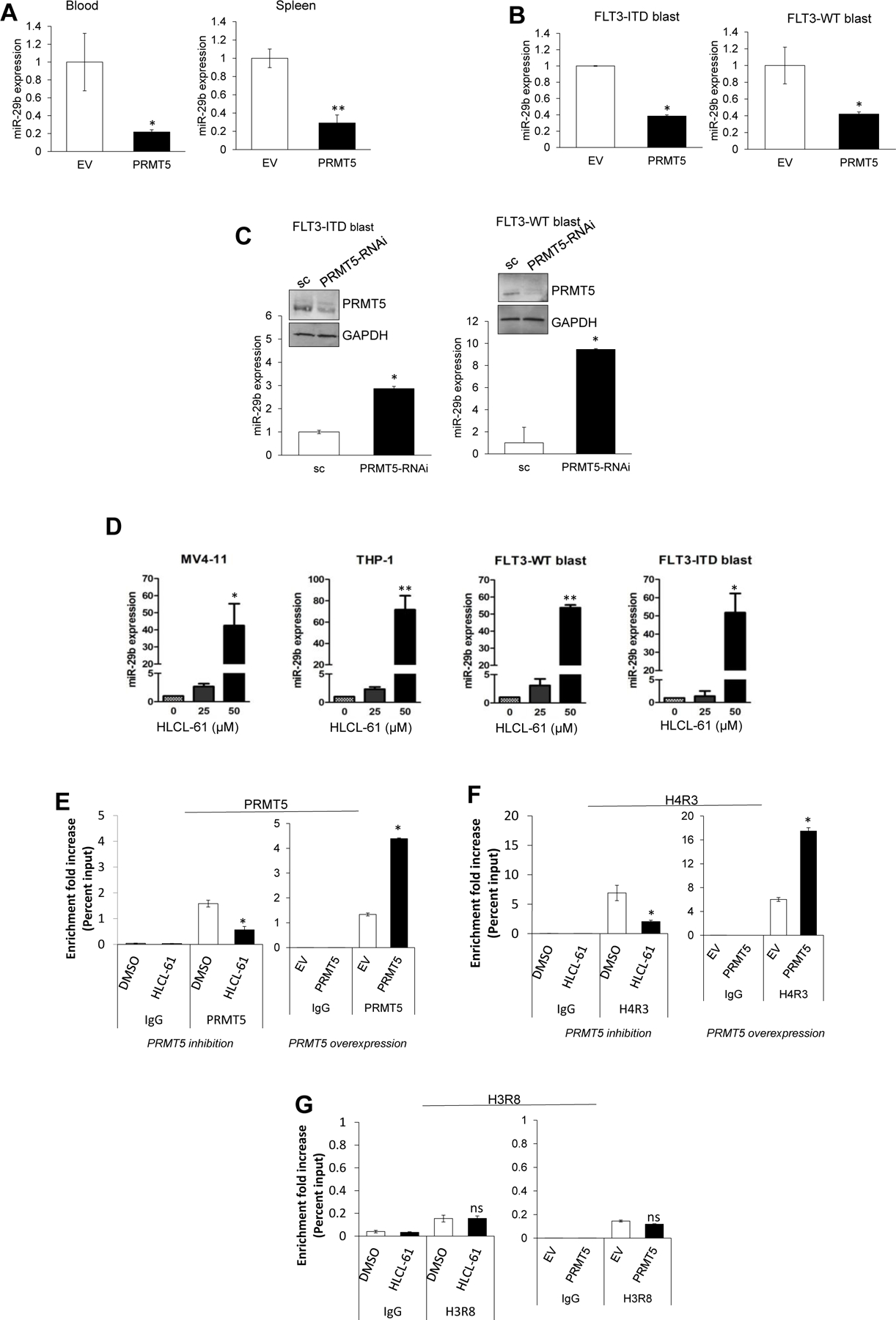
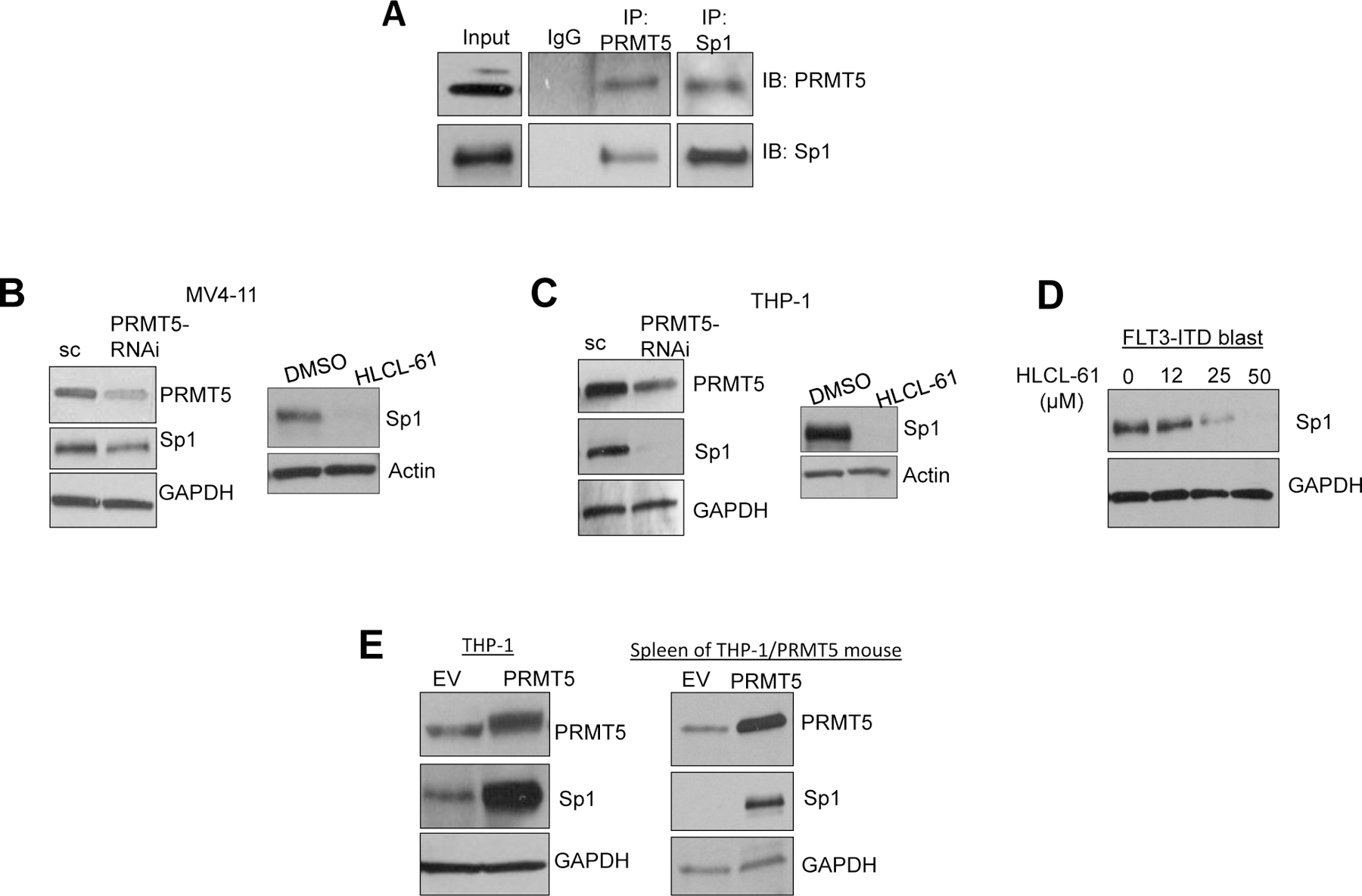
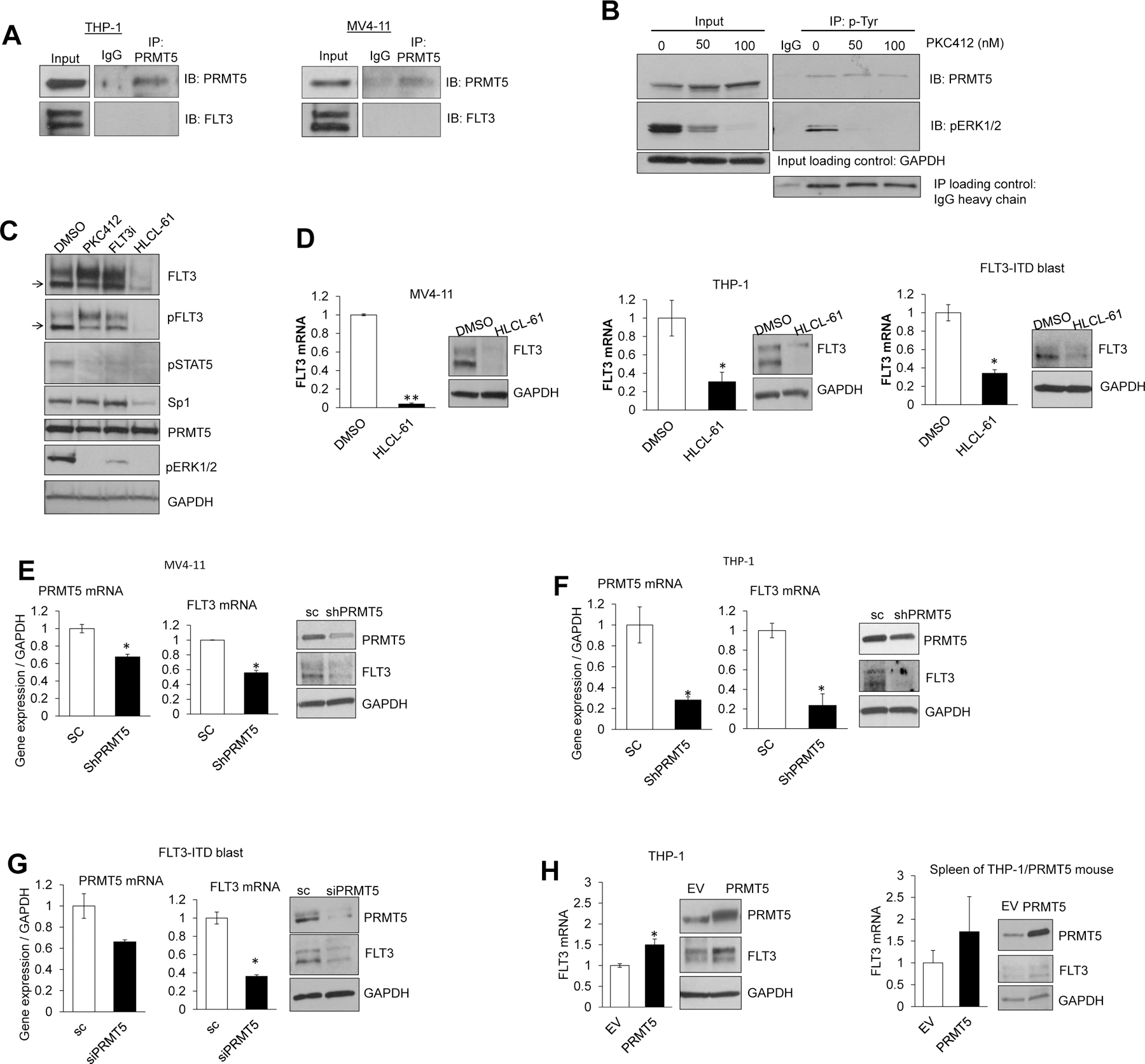
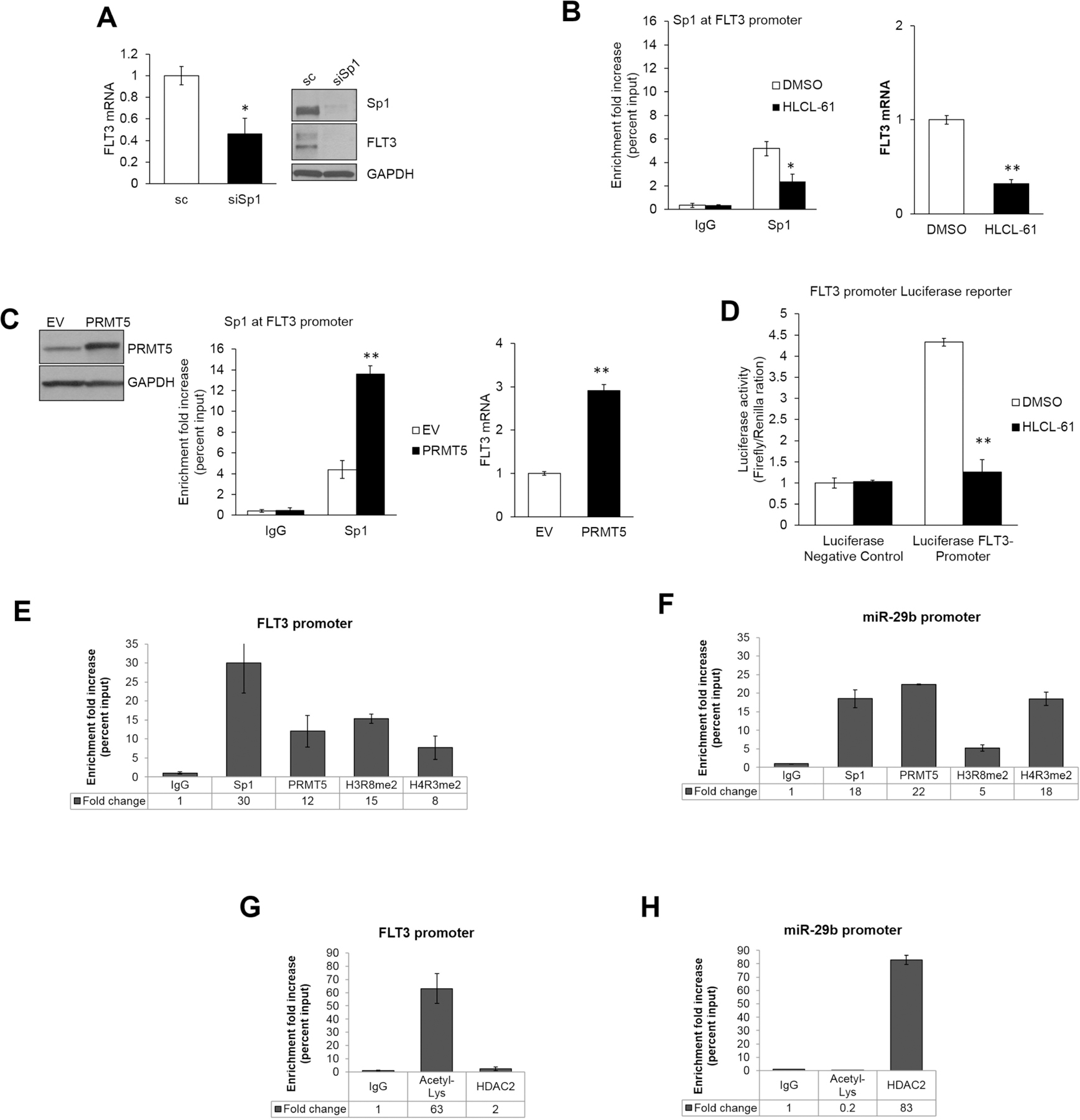
Changes in the enzymatic activity of protein arginine methyltransferase (PRMT) 5 have been associated with cancer; however, the protein's role in acute myeloid leukemia (AML) has not been fully evaluated. Here, we show that increased PRMT5 activity enhanced AML growth in vitro and in vivo while PRMT5 downregulation reduced it. In AML cells, PRMT5 interacted with Sp1 in a transcription repressor complex and silenced miR-29b preferentially via dimethylation of histone 4 arginine residue H4R3. As Sp1 is also a bona fide target of miR-29b, the miR silencing resulted in increased Sp1. This event in turn led to transcription activation of FLT3, a gene that encodes a receptor tyrosine kinase. Inhibition of PRMT5 via sh/siRNA or a first-in-class small-molecule inhibitor (HLCL-61) resulted in significantly increased expression of miR-29b and consequent suppression of Sp1 and FLT3 in AML cells. As a result, significant antileukemic activity was achieved. Collectively, our data support a novel leukemogenic mechanism in AML where PRMT5 mediates both silencing and transcription of genes that participate in a 'yin-yang' functional network supporting leukemia growth. As FLT3 is often mutated in AML and pharmacologic inhibition of PRMT5 appears feasible, the PRMT5-miR-29b-FLT3 network should be further explored as a novel therapeutic target for AML.
蛋白质精氨酸甲基转移酶(PRMT)5的酶活性变化与癌症相关;然而,该蛋白在急性髓系白血病(AML)中的作用尚未得到充分评估。在此,我们表明PRMT5活性增加会增强AML在体外和体内的生长,而PRMT5下调则会使其生长减缓。在AML细胞中,PRMT5在转录抑制复合物中与Sp1相互作用,并通过组蛋白4精氨酸残基H4R3的二甲基化优先沉默miR-29b。由于Sp1也是miR-29b的真正靶点,miR沉默导致Sp1增加。这一事件进而导致FLT3的转录激活,FLT3是一种编码受体酪氨酸激酶的基因。通过sh/siRNA或一类首创的小分子抑制剂(HLCL-61)抑制PRMT5会导致AML细胞中miR-29b的表达显著增加,从而抑制Sp1和FLT3。结果,实现了显著的抗白血病活性。总体而言,我们的数据支持AML中一种新的白血病发生机制,即PRMT5介导参与支持白血病生长的“阴阳”功能网络的基因的沉默和转录。由于FLT3在AML中经常发生突变,并且对PRMT5的药物抑制似乎可行,PRMT5-miR-29b-FLT3网络应作为AML的新治疗靶点进一步探索。