Lim Lee Ling, Kitan Normayah, Paramasivam Sharmila Sunita, Ratnasingam Jeyakantha, Ibrahim Luqman, Chan Siew Pheng, Tan Alexander Tong Boon, Vethakkan Shireene Ratna
Division of Endocrinology, Department of Internal Medicine, University of Malaya Medical Center, Lembah Pantai, 59100, Kuala Lumpur, Malaysia.
Division of Endocrine Surgery, Department of Surgery, Putrajaya Hospital, Putrajaya, Malaysia.
J Med Case Rep. 2015 Dec 1;9:277. doi: 10.1186/s13256-015-0757-7.
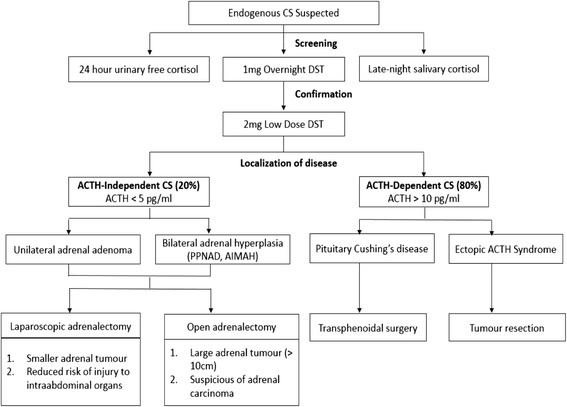
Determining the etiology of Cushing's syndrome is very challenging to endocrinologists, with most of the difficulty arising from subtype differentiation of adrenocorticotropic hormone-dependent Cushing's syndrome. We present the pitfalls of evaluating a rare cause of adrenocorticotropic hormone-independent Cushing's syndrome in the transition period between adolescence and adulthood.
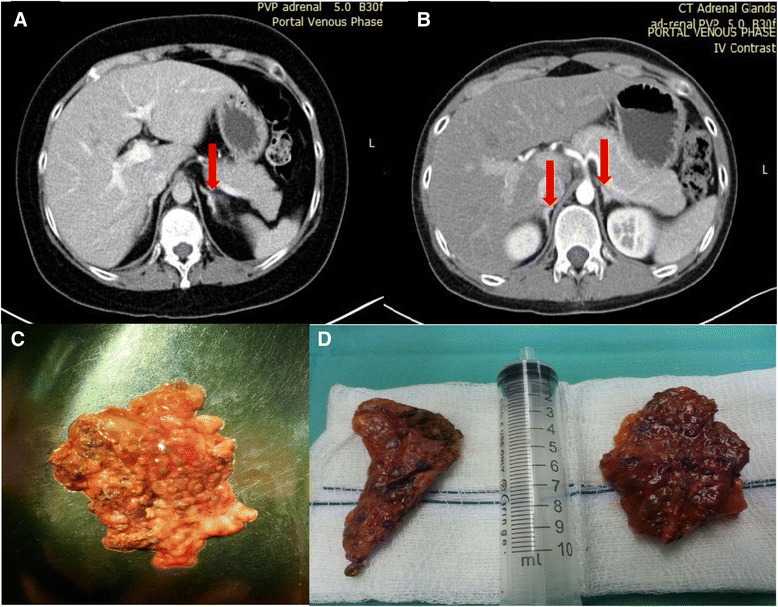
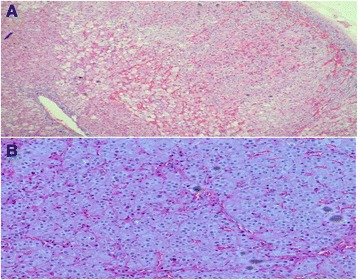
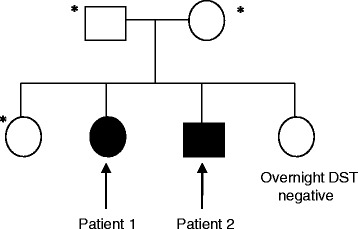
A sibling pair with familial isolated primary pigmented nodular adrenocortical disease is described. The index case, a 20-year-old Chinese woman, presented with premenopausal osteoporosis with T12 compression fracture and young hypertension. Biochemical analysis confirmed adrenocorticotropic hormone-independent Cushing's syndrome (elevated 0800 h plasma cortisol 808 nmol/L with suppressed adrenocorticotropic hormone level <5 pg/ml). Computed tomography of her adrenal glands revealed a 0.7-cm left adrenal hypodense nodule. After a left adrenalectomy, she had residual hypercortisolism (progressive weight gain, new T10 compression fracture, and not glucocorticoid-dependent postoperatively). Completion of contralateral adrenalectomy was performed upon recognition of typical histologic characteristics of primary pigmented nodular adrenocortical disease found in an initial left adrenalectomy specimen. Similarly, her younger brother developed adrenocorticotropic hormone-independent Cushing's syndrome at age 18 years, with typical cushingoid habitus, but no osteoporosis or hypertension. His adrenal computed tomographic scans showed micronodularities over bilateral adrenal glands. He was successfully treated with bilateral adrenalectomy. Screening for Carney's complex and PRKAR1A gene mutation was negative. Signs and symptoms of Cushing's syndrome resolved after bilateral adrenalectomy for both patients. They were placed on lifelong glucocorticoid and mineralocorticoid replacement therapy and long-term surveillance for Carney's complex.
The cases of these two patients illustrate the difficulties involved in diagnosing primary pigmented nodular adrenocortical disease, a variant of adrenocorticotropic hormone-independent Cushing's syndrome that is managed with bilateral adrenalectomy. A high index of suspicion for this disease is needed, especially in adolescents with adrenocorticotropic hormone-independent Cushing's syndrome who have a significant family history, features of Carney's complex, and no resolution of Cushing's syndrome after unilateral adrenalectomy. Patients with primary pigmented nodular adrenocortical disease can either have bilateral/multiple adrenal nodules or normal adrenal glands visualized by computed tomography. Long-term surveillance is imperative in patients with confirmed Carney's complex and in those who have not undergone complete genetic testing to exclude this hereditary disorder.
对于内分泌科医生而言,确定库欣综合征的病因极具挑战性,其中大部分困难源于促肾上腺皮质激素依赖性库欣综合征的亚型分化。我们介绍了在青少年到成年的过渡期评估一种罕见的促肾上腺皮质激素非依赖性库欣综合征病因时的陷阱。
描述了一对患有家族性孤立性原发性色素沉着性结节性肾上腺皮质疾病的兄弟姐妹。索引病例是一名20岁的中国女性,表现为绝经前骨质疏松伴T12压缩性骨折和青年高血压。生化分析证实为促肾上腺皮质激素非依赖性库欣综合征(上午8点血浆皮质醇升高至808 nmol/L,促肾上腺皮质激素水平抑制至<5 pg/ml)。肾上腺计算机断层扫描显示左侧肾上腺有一个0.7厘米的低密度结节。左侧肾上腺切除术后,她仍有皮质醇增多症残留(体重逐渐增加、新发T10压缩性骨折且术后不依赖糖皮质激素)。在认识到最初左侧肾上腺切除标本中发现的原发性色素沉着性结节性肾上腺皮质疾病的典型组织学特征后,进行了对侧肾上腺切除术。同样,她的弟弟在18岁时患上促肾上腺皮质激素非依赖性库欣综合征,具有典型的库欣样外貌,但无骨质疏松或高血压。他的肾上腺计算机断层扫描显示双侧肾上腺有小结节。他通过双侧肾上腺切除术成功治愈。对卡尼综合征和PRKAR1A基因突变的筛查均为阴性。两名患者双侧肾上腺切除术后库欣综合征的体征和症状均得到缓解。他们开始接受终身糖皮质激素和盐皮质激素替代治疗,并对卡尼综合征进行长期监测。
这两名患者的病例说明了诊断原发性色素沉着性结节性肾上腺皮质疾病的困难,这是一种促肾上腺皮质激素非依赖性库欣综合征的变体,通过双侧肾上腺切除术进行治疗。对于这种疾病需要高度怀疑,特别是在有明显家族史、具有卡尼综合征特征且单侧肾上腺切除术后库欣综合征未缓解的促肾上腺皮质激素非依赖性库欣综合征青少年患者中。原发性色素沉着性结节性肾上腺皮质疾病患者的计算机断层扫描可能显示双侧/多发性肾上腺结节或肾上腺正常。对于确诊为卡尼综合征的患者以及未进行全面基因检测以排除这种遗传性疾病的患者,必须进行长期监测。