Lee Sook-Jeong, Seo Bo-Ra, Koh Jae-Young
Neural Injury Research Center, Asan Institute for Life Sciences, University of Ulsan College of Medicine, Seoul, South Korea.
Present address: Department of New Drug Discovery and Development, Chungnam National University, 99 Daehak-ro, Yuseong, Daejeon, 34134, South Korea.
Mol Brain. 2015 Dec 4;8(1):84. doi: 10.1186/s13041-015-0173-3.
Astrocytes may play important roles in the pathogenesis of Alzheimer's disease (AD) by clearing extracellular amyloid beta (Aβ) through endocytosis and degradation. We recently showed that metallothionein 3 (Mt3), a zinc-binding metallothionein that is enriched in the central nervous system, contributes to actin polymerization in astrocytes. Because actin is likely involved in the endocytosis of Aβ, we investigated the possible role of Mt3 in Aβ endocytosis by cortical astrocytes in this study.
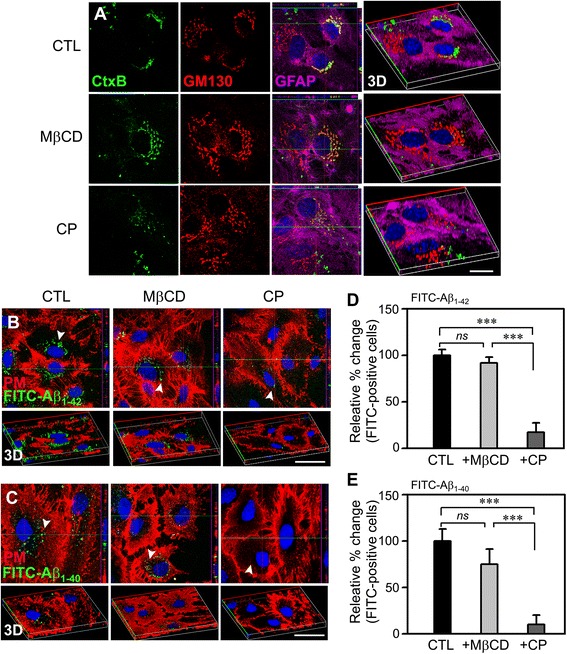
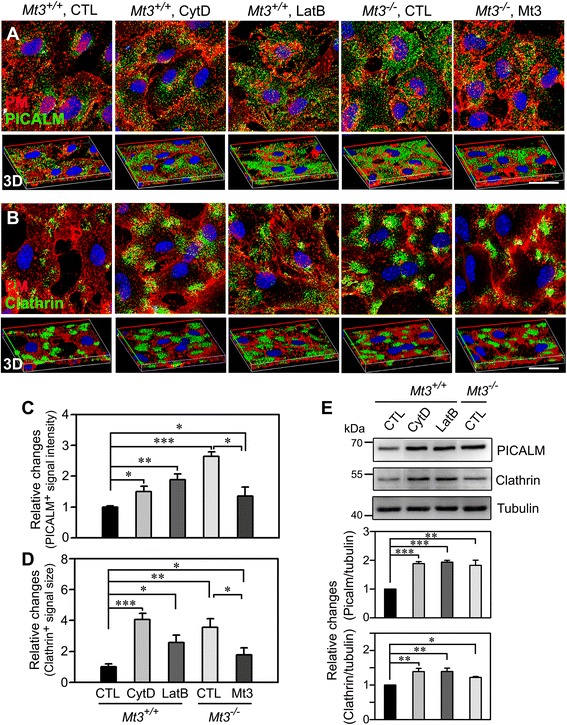
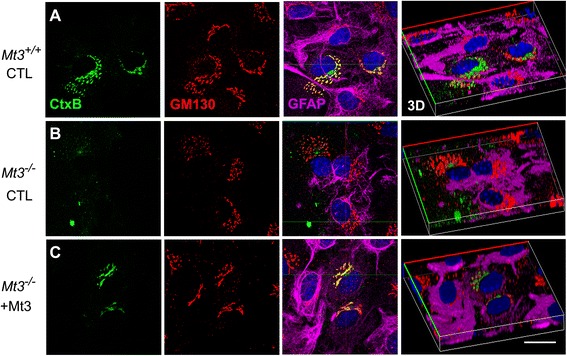
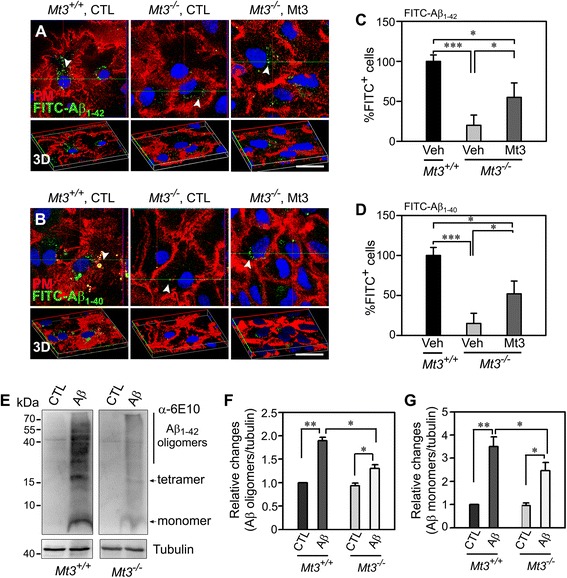
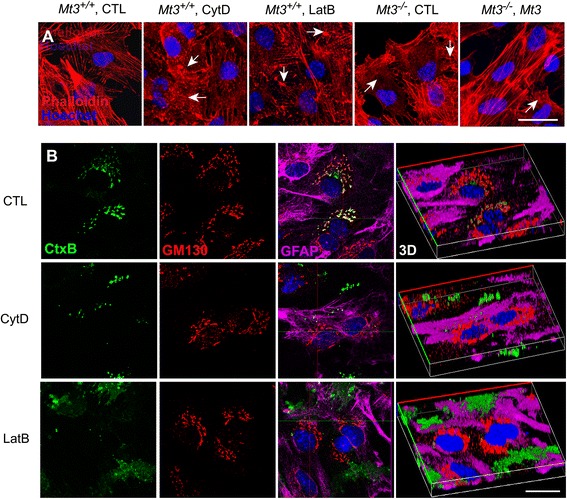
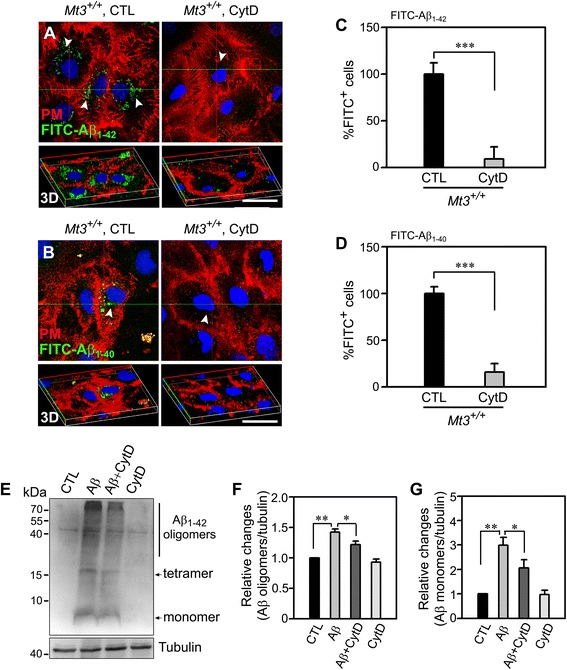
To assess the route of Aβ uptake, we exposed cultured astrocytes to fluorescently labeled Aβ1-40 or Aβ1-42 together with chloropromazine (CP) or methyl-beta-cyclodextrin (MβCD), inhibitors of clathrin- and caveolin-dependent endocytosis, respectively. CP treatment almost completely blocked Aβ1-40 and Aβ1-42 endocytosis, whereas exposure to MβCD had no significant effect. Actin disruption with cytochalasin D (CytD) or latrunculin B also completely blocked Aβ1-40 and Aβ1-42 endocytosis. Because the absence of Mt3 also results in actin disruption, we examined Aβ1-40 and Aβ1-42 uptake and expression in Mt3 (-/-) astrocytes. Compared with wild-type (WT) cells, Mt3 (-/-) cells exhibited markedly reduced Aβ1-40 and Aβ1-42 endocytosis and expression of Aβ1-42 monomers and oligomers. A similar reduction was observed in CytD-treated WT cells. Finally, actin disruption and Mt3 knockout each increased the overall levels of clathrin and the associated protein phosphatidylinositol-binding clathrin assembly protein (PICALM) in astrocytes.
Our results suggest that the absence of Mt3 reduces Aβ uptake in astrocytes through an abnormality in actin polymerization. In light of evidence that Mt3 is downregulated in AD, our findings indicate that this mechanism may contribute to the extracellular accumulation of Aβ in this disease.
星形胶质细胞可能通过内吞作用和降解清除细胞外淀粉样β蛋白(Aβ),在阿尔茨海默病(AD)的发病机制中发挥重要作用。我们最近发现,金属硫蛋白3(Mt3)是一种在中枢神经系统中富集的锌结合金属硫蛋白,它有助于星形胶质细胞中的肌动蛋白聚合。由于肌动蛋白可能参与Aβ的内吞作用,我们在本研究中调查了Mt3在皮质星形胶质细胞Aβ内吞作用中的可能作用。
为了评估Aβ摄取的途径,我们将培养的星形胶质细胞暴露于荧光标记的Aβ1-40或Aβ1-42中,同时分别加入氯丙嗪(CP)或甲基-β-环糊精(MβCD),它们分别是网格蛋白依赖性内吞作用和小窝蛋白依赖性内吞作用的抑制剂。CP处理几乎完全阻断了Aβ1-40和Aβ1-42的内吞作用,而暴露于MβCD则没有显著影响。用细胞松弛素D(CytD)或拉春库林B破坏肌动蛋白也完全阻断了Aβ1-40和Aβ1-42的内吞作用。由于Mt3的缺失也会导致肌动蛋白破坏,我们检测了Mt3(-/-)星形胶质细胞中Aβ1-40和Aβ1-42的摄取及表达。与野生型(WT)细胞相比,Mt3(-/-)细胞中Aβ1-40和Aβ1-42的内吞作用以及Aβ1-42单体和寡聚体的表达明显降低。在CytD处理的WT细胞中也观察到了类似的降低。最后,肌动蛋白破坏和Mt3基因敲除均增加了星形胶质细胞中网格蛋白和相关蛋白磷脂酰肌醇结合网格蛋白组装蛋白(PICALM)的总体水平。
我们的结果表明,Mt3的缺失通过肌动蛋白聚合异常降低了星形胶质细胞对Aβ的摄取。鉴于有证据表明Mt3在AD中表达下调,我们的发现表明这种机制可能导致该疾病中Aβ的细胞外积累。